



INTRODUCTION
The Australian National University has a long history of radiocarbon dating. The first radiocarbon laboratory, based on gas proportional counting, was set up in 1965 by Henry Polach (laboratory code ANU#) and continued to operate until 2003. Between 2002 and 2014 the 14 UD Pelletron accelerator was occasionally used for radiocarbon dating (laboratory code ANUA-#). This AMS was used for radiocarbon prior to 2002, but ANUA-# numbers were not assigned. In 2007 an NEC Single Stage AMS was installed and used solely for 14C analysis (laboratory code S-ANU#) (Fallon et al. Reference Fallon, Fifield and Chappell2010). This paper outlines the sample preparation methods used by the S-ANU laboratory. These are regularly reviewed and updated, and this paper describes methods used since 2011.
Samples submitted to the laboratory are derived from archaeological, forensic, palaeoenvironmental and oceanographic contexts, spanning a range of carbonates, organics and water (Figure 1). Most samples submitted to the laboratory are from regions with exceptionally poor organic preservation. Around a third of fossil samples are from Australia, a third from Southeast Asia, and a fifth from the Pacific. This paper aims to share experience obtained working on samples from challenging regions, and comments on quality assurance indicators and preservation of commonly dated materials.

Figure 1 Materials submitted to the S-ANU laboratory since 2011. The category “other” includes laboratory tests of reagents and conservation treatments found on samples, as well some of the rarer sample types such as textiles, beeswax, paper and industrial products such as additives in the wine industry and fuels.
STORAGE OF SAMPLES AND DATABASING
All Australian human remains are returned to the sample submitter. Other samples are returned at the submitter’s request, or where required by laboratory staff. Samples are stored in plastic bags and acid-free cardboard boxes when not returned. Aluminum foil is avoided in most cases as this degrades when exposed to high or low pH. Records are retained in paper format and in a Filemaker Pro™ database.
GENERAL CONDITIONS
All chemicals used for chemical pretreatment are at least AR grade. Solvents are HPLC grade (Lichrosolv, DCM stabliser amylene), and type 1 (ultrapure) water is used. In February 2021 a MilliQ™ system (Synergy UV) was replaced with a Sartorious™ Arium™ Comfort system. All glassware is cleaned using a series of 0.1% Decon™, 0.01 M HCl and ultrapure water washes (>1 hour), before combustion at 400ºC/3 hours. Ezee-Filters™ are used to help remove the liquid from most samples, and cleaned by rinsing and ultrasonication (30 minutes) in ultrapure water. Samples are freeze-dried using a FreezeZone™ 2.5 freeze-drier connected to an oil-free vacuum pump.
ISOTOPE RATIO MASS SPECTROMETRY
δ13C, δ15N, and δ18O are measured on a Sercon 20-22 isotope ratio mass spectrometer connected to either an ANCA GSL or a carbonate device operating in continuous flow mode. Standards are matched to the sample expected %C, %N and isotope value, and normalized using a multi-point scaling process (Paul et al. Reference Paul, Skrzypek and Fórizs2007). For example, collagen samples are normally measured against an in-house gelatin reference, scaled using USGS40, USGS65, and IAEA-C6, and data accuracy is assessed with IAEA-600 and USGS61. Since August 2014, samples and standards have been weighed using a Sartorius™ Cubi™ microbalance. Typical reproducibility is < 0.2‰ for δ13C and δ15N on gas from the ANCA GSL, and 0.06‰ for δ13C and 0.1‰ for δ18O on gas produced on the carbonate device.
PHYSICAL PREPARATION
Samples are inspected, under a binocular microscope where required, and cleaned with a scalpel or handheld Dremel™ drill (bone and shell) to remove rootlets, sediment and degraded material. All attempts are made to sample single entities. Most are crushed to around 2 mm particle size using an agate pestle and mortar or a scalpel to increase surface area for cleaning, as described below. To limit the potential for cross-contamination, samples are not sieved and a range of particle sizes are present.
SOLVENT WASHING
Where samples have visible, or suspected glue or conservation treatments, a series of solvent washes is applied to suit the contaminant present (Brock et al. Reference Brock, Higham, Ditchfield and Ramsey2010; Dee et al. Reference Dee, Brock, Bowles and Ramsey2011; O’Regan et al. Reference O’Regan, Petchey, Wood, Mcalister, Bradshaw and Holdaway2019). These may not fully remove the conservation treatment (Brock et al. Reference Brock, Dee, Hughes, Snoeck, Staff and Bronk Ramsey2018), and conserved materials are avoided where possible. In September 2019 the laboratory changed from using chloroform to dichloromethane for safety reasons. The glue or conservation treatment is rarely known, and the most common solvent wash applied is acetone, methanol and dichloromethane (1 hour each, replacing if becoming colored). Acetone and methanol can be heated to 40ºC and/or ultrasonicated where the sample is robust. This series of washes is also used on young (< about 500 years) wood to remove resins. Young (< about 500 years) or very well preserved (visual inspection) bone is defatted in a 2:1 mixture of dichloromethane and methanol (1 hour, room temperature, replacing the solution until no color is removed) (Folch et al. Reference Folch, Lees and Stanley1957). This method is more effective at removing low levels of lipids (<2%) than the Bligh and Dyer method (Iverson et al. Reference Iverson, Lang and Cooper2001).
PLANT MATERIAL
Charred Plant Material
2–20 mg of charcoal is cleaned under a bifocal microscope to remove sediments and soft or vitreous degraded material and cut or crushed to 1–2 mm diameter fragments. Most samples are pretreated using an acid-base-acid (ABA) procedure consisting of HCl, NaOH and HCl washes (Table 1). After each treatment samples are rinsed at least 3 times in ultrapure water, or until the water remains colorless.
Table 1 Pretreatment conditions in pretreatment of burnt and unburnt plant material. Between each treatment, the sample is washed at least 3 times in ultrapure water, or until the water remains colorless. RT refers to room temperature.

Large (>50 mg) charcoal samples thought to be more than 30 ka are subjected to ABOx-SC (Bird et al. Reference Bird, Ayliffe, Fifield, Tumey, Cresswell, Barrows and David1999; Wood et al. Reference Wood, Jacobs, Vannieuwenhuyse, Balme, O’Connor and Whitau2016b). Between 50–100 mg charcoal is prepared as described for the ABA procedure, and subjected to treatment with 6M HCl (1 hour, room temperature), 2M NaOH (30 minutes, room temperature, solution replaced until it remains colorless) and a solution of 0.1M K2Cr2O7 in 2M H2SO4 (60ºC, 20 hours, screw top tube). After each treatment the charcoal is rinsed at least 3 times in ultrapure water, or until the water remains colorless. Rather than a stepped combustion described by (Bird et al. Reference Bird, Ayliffe, Fifield, Tumey, Cresswell, Barrows and David1999), charcoal is precombusted at 600ºC for 2 hours and the CO2 generated is discarded. To do this, 10–20 mg of cleaned charcoal is placed in a quartz tube sealed with a quartz wool plug and evacuated to <3×10–3 Torr on a graphitization rig as described below, and 730 Torr (1 atmosphere) ultra high purity (>99.995) O2 is added. Any charcoal remaining is recovered from the quartz tube.
When samples >30 ka are dated, both ABA and ABOx-SC are undertaken on several charcoal fragments from a given site where possible. Contaminants are assumed insignificant if results are indistinguishable, and the remaining samples are pretreated with ABA. Where different, remaining samples are treated with ABOx-SC.
Charcoal ABA yields range considerably (Figure 2a). Typically, at least 50% of a charcoal sample will survive the ABA pretreatment protocol, but in 20% of cases more than 90% of a charcoal sample was lost. In tropical locations, preservation can be minimal in relatively young sites, especially where sediments have a high pH (e.g. Loc Giang; Piper et al. Reference Piper, Nguyen, Tran, Wood, Cobo Castillo, Weisskopf, Campos, Dang, Sarjeant, Mijares, Oxenham and Bellwood2017). Charcoal quality is assessed by %C, measured volumetrically while collecting CO2 for graphitization. %C of charcoal is dependent on a range of factors, including species and temperature of pyrolysis, with %C typically ranging between 50 and 85 %C (Ascough et al. Reference Ascough, Bird, Francis and Lebl2011, Braadbaart and Poole Reference Braadbaart and Poole2008). %C of fossil charcoal can decrease through diagenesis via oxidation (Ascough et al. Reference Ascough, Bird, Francis and Lebl2011), and because fine sediment can accumulate within vessels and be concentrated if degraded charcoal is dissolved in pretreatment (Rebollo et al. Reference Rebollo, Weiner, Brock, Meignen, Goldberg, Belfer-Cohen, Bar-Yosef and Boaretto2011). It is therefore unsurprising that %C is not related to % yield (Figure 2b). While oxidation is not always related to the presence of exogenous carbon (Ascough et al. Reference Ascough, Bird, Francis and Lebl2011), sediment particles can introduce carbon of very different age to the charcoal (Rebollo et al. Reference Rebollo, Weiner, Brock, Meignen, Goldberg, Belfer-Cohen, Bar-Yosef and Boaretto2011). Therefore, the S-ANU laboratory will normally only date charcoal containing more than 50% C. 7% of charcoal graphitized between 2011 and 2021 did not meet this criterion. Where charcoal with < 50% C is dated, the laboratory provides a warning to the sample submitter.

Figure 2 Success of the ABA pretreatment applied to charcoal since 2011 (a) % yield (b) comparison of %C and % yield.
Unburnt Plant Material
Plant materials are physically cleaned and inspected as described for charcoal, before being crushed or cut to ca. 1 mm pieces, or in the case of wood, into thin shavings. Samples are treated using one of several acid-base-acid procedures based on sample age and fragility (assessed visually) (Table 1). %C is used as a quality assurance measure. When outside of 40–50% C, a note is attached to the date report to indicate that the accuracy of the date may be low.
SEDIMENT
Sediment samples are only dated after close consultation with the sample submitter, and only from palaeoenvironmental contexts where no other potential samples are present. In most cases two ages are obtained (e.g., ABA residue 14C001A and base soluble humics 14C001G; Table 1) to assess the age difference of different sediment components, and thus aid interpretation.
HUMAN AND FAUNAL REMAINS
Bone and Dentine—Fossil
Where bones are visibly degraded or preservation is expected to be poor, collagen preservation is assessed with %N (Brock et al. Reference Brock, Wood, Higham, Ditchfield, Bayliss and Ramsey2012). The bone surface is removed with a drill, and 2–5 mg of bone is taken for measurement by EA-IRMS. More than one %N sample is taken from a bone/ skeleton where preservation is expected to be variable, for example from dentine and bone in a mandible, from different skeletal elements in an inhumation, or across a large bone (Calo et al. Reference Calo, Prasetyo, Bellwood, Lankton, Gratuze, Pryce, Reinecke, Leusch, Schenk, Wood, Bawono, Kompiang Gede, Citha Yuliati, Fenner, Reepmeyer, Castillo and Carter2015; Jacob et al. Reference Jacob, Querci, Caparros, Barroso Ruiz, Higham and Deviese2018). All bones containing > 0.7% N are pretreated. Bones containing more than 0.2% N are pretreated after consultation with the sample submitter on the understanding that the likelihood of producing sufficient collagen is <70%. Samples with <0.2% N, the theoretical N content required to yield 1% collagen, are not pretreated.
To radiocarbon date bone, the surface and soft degraded material is removed with a drill. Samples are either ground with a hand pestle and mortar or crushed in a bone press to ca. 1 mm pieces, or drilled. Collagen yields are higher when ground as previously observed (Fewlass et al. Reference Fewlass, Tuna, Fagault, Hublin, Kromer, Bard and Talamo2019), and crushing is favored unless damage needs to be minimized, for example when samples are culturally or morphologically significant. When ground, 6.8 ± 1.2wt.% collagen (n=7) is extracted from the Zaglik bone standard, and when drilled 3.1 ± 1.7% (n=9) is extracted (starting weight >400 mg).
Collagen is extracted and purified using an ultrafiltration method, with conditions based on (Brock et al. Reference Brock, Higham, Ditchfield and Ramsey2010; Ramsey et al. Reference Ramsey, Higham, Bowles and Hedges2004). Bone is demineralized using four treatments of ca. 20 mL 0.5M HCl. The first two are at room temperature for 1–2 hours, the third overnight at 5°C and the fourth for 1–2 hours at room temperature. Samples are then submerged in ca. 20 mL 0.1M NaOH for 30 minutes to remove humics, replacing where the solution is colored, and finally 0.5M HCl at room temperature for 1 hour. After each treatment, the sample is washed three times in ultrapure water. A centrifuge and glass pipettes are used to aid removal of the supernatant. The crude collagen is gelatinized in 10 mL 0.001M HCl (pH3) at 70ºC for 20 hours in 35-mL screw top tubes in a Binder™ oven, before filtration with an Ezee-Filter™ to remove larger insoluble contaminants. Gelatin is ultrafiltered using Vivaspin™ VS15 Turbo 30kDa MWCO ultrafilters, which have a polyethersulphone membrane, until <1 mL solution remains in the ultrafilter, and the >30kDa fraction is collected and freeze-dried. Ultrafilters work on a molecular scale, with complex electrostatic interactions between the molecules in solution, and the solution and membrane. Although some small proteins are removed, the process produces a sample with a range of proteins, including collagen, of different sizes (Brock et al. Reference Brock, Geoghegan, Thomas, Jurkschat and Higham2013a).
Ultrafilters are cleaned prior to use by centrifuging ca. 15 mL ultrapure water twice (around 10 minutes each), 1-hour ultrasonication in ultrapure water, followed by centrifuging ca. 15 mL ultrapure water a further three times to remove the humectant glycerol. To monitor possible fluctuations in age, one sample of glycerol per ultrafilter batch is collected from the washing process (Brock et al. Reference Brock, Higham and Ramsey2013b). Glycerol has been modern in all batches of ultrafilter used by the facility. A bone specific background is subtracted (Wood et al. Reference Wood, Bronk Ramsey and Higham2010; Fallon et al. in prep).
Collagen preservation is assessed with collagen yield (wt.%), %C and C:N ratio (van Klinken Reference Van Klinken1999). Various cut-off values have been used by different radiocarbon laboratories, and between the radiocarbon and stable isotope fields. To date, the ANU laboratory has accepted samples containing more than 1 wt.% collagen, 30% C, and having a C:N between 2.9 and 3.4, adapted from the protocol used at ORAU (Brock et al. Reference Brock, Higham, Ditchfield and Ramsey2010), derived from van Klinken (Reference Van Klinken1999). However, measurements for both %C and C:N over the last 7 years (Figure 3b, c) are more consistent than those published by van Klinken (Reference Van Klinken1999). This is most likely due to the use of ultrafiltration as well as improvements in IRMS and use of a micro-balance when weighing samples, and implies the acceptance criteria need to be re-evaluated.
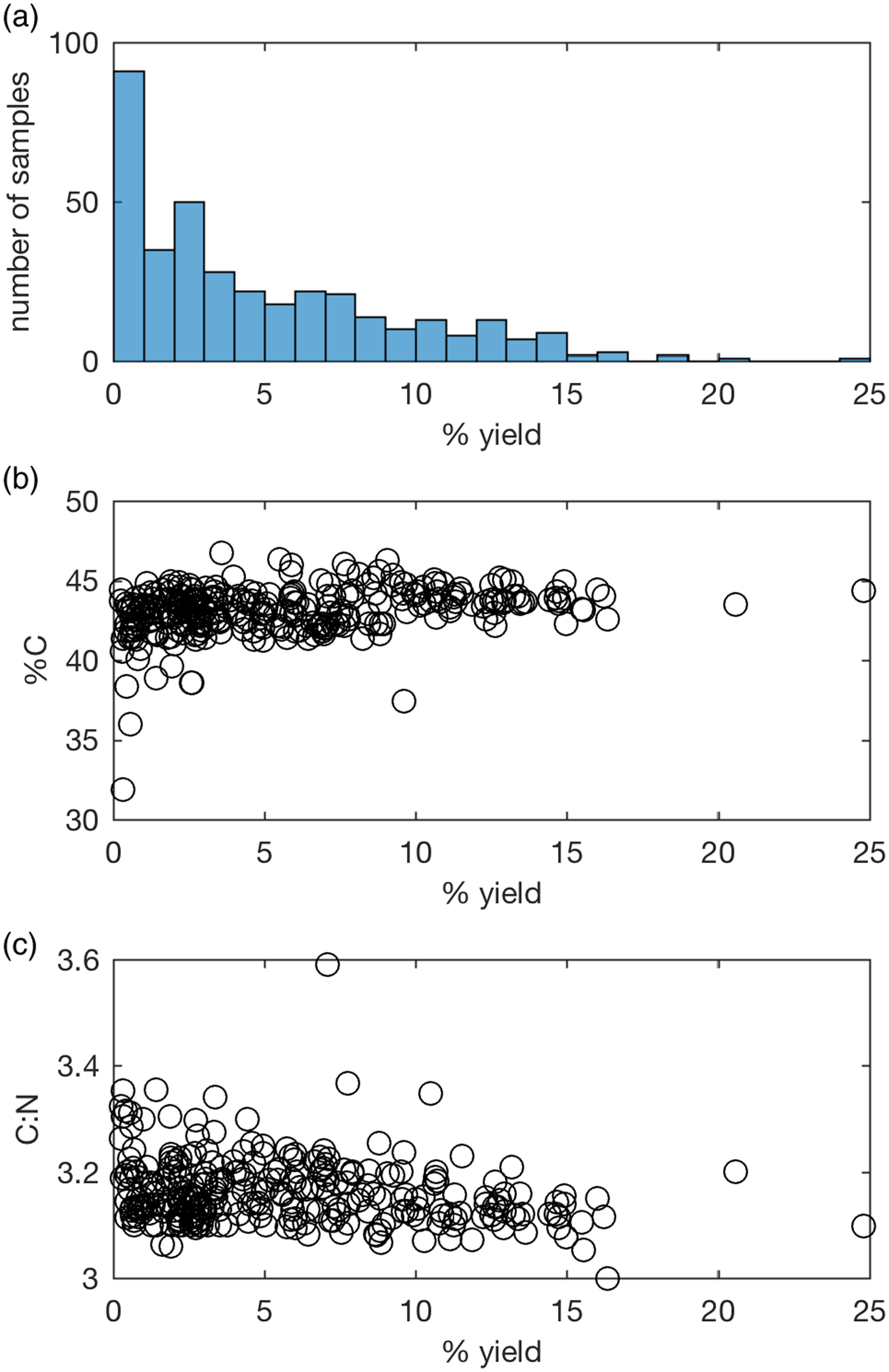
Figure 3 Collagen quality indicators of samples pretreated since August 2014: (a) % collagen yield, (b) %C, (c) C:N.
For samples containing >1% collagen, and excluding a group of Egyptian mummies expected to be affected by resins (Wasef et al. Reference Wasef, Wood, El Merghani, Ikram, Curtis, Holland, Willerslev, Millar and Lambert2015), %C averages 43.3 ± 1.2 and C:N 3.15 ± 0.05 (n=250). From May 2021 the laboratory has accepted samples within 3 standard deviations of the average, i.e., 39.7–46.9% C and C:N ratio of 3.00–3.30. Although the C:N ratio is a relatively insensitive indicator for contaminants, it is often 3.4 or higher when dates on ultrafiltered collagen are younger than dates on hydroxyproline, justifying this tighter criteria (Marom et al. Reference Marom, McCullagh, Higham, Sinitsyn and Hedges2012; Devièse et al. Reference Devièse, Abrams, Hajdinjak, Pirson, De Groote, Di Modica, Toussaint, Fischer, Comeskey, Spindler, Meyer, Semal and Higham2021; Spindler et al. Reference Spindler, Comeskey, Chabai, Uthmeier, Buckley, Devièse and Higham2021). Although %C of samples with low collagen yields are more likely to be lower than better preserved bone, most fall within the range of bone where >1% collagen was obtained. Therefore, samples containing 0.5–1% collagen will be dated where EA-IRMS values are acceptable, substantially extending the number of datable samples (Figure 3a).
δ13C and δ15N are also measured, but their use as indicators for preservation is somewhat limited within Australia and South East Asia as there is considerable geographic variability due to variation in plant composition and potential evapotranspiration (Fraser Reference Fraser2005; Murphy Reference Murphy2006) and, in many regions, minimal baseline data for most species.
Bone and Dentine—Forensic
A simplified collagen extraction is undertaken for very recent (post-1950) samples as little contamination is expected, and the effect of young contaminants is minimal. A small (100–200 mg) sample of bone/dentine is defatted as described above, and collagen extracted by demineralization in HCl, gelatinization and Ezee filtration, as described for ancient samples.
Cremated Bone
Bone burnt or charred at low temperature cannot be radiocarbon dated as the protein is damaged. However, when heated to more than 600ºC so that the bone is grey or white, bone apatite recrystallizes into larger crystals that are more stable and less prone to diagenesis or contamination than the mineral phase of unburnt bone, and can be dated (Lanting et al. Reference Lanting, Aerts-Bijma and van der Plicht2001). The carbonate in cremated or calcined bone is a mixture of endogenous carbon from the animal/human and carbonate from the fuel and thus can have an inbuilt age (Hüls et al. Reference Hüls, Erlenkeuser, Nadeau, Grootes and Andersen2010; van Strydonck et al. Reference Van Strydonck, Boudin and De Mulder2010; Snoeck et al. Reference Snoeck, Staff and Brock2016).
After thorough removal of the surface with a drill and grinding to <1 mm, around 400 mg cremated bone is reacted with 1 M acetic acid (1 mL/ 50 mg bone) for 20 hours in a Vacutainer™ evacuated to <1x10-2 Torr and rinsed 5 times with ultrapure water. This treatment removes 5–10% of the bone.
To assess whether the sample is calcined, 1–1.5 mg bone is ground with 200–300 mg KBr and pressed into a pellet under vacuum for FTIR analysis. The average of 100 transmission spectra are recorded on a Bruker Tensor 27 between –400 and –4000 cm–1, at a resolution of 4 cm–1. The infra-red splitting factor (IRSF) provides an indication of crystal order, with values above 4 indicative of partial recrystallization and values around 7 indicative of complete recrystallization (Olsen et al. Reference Olsen, Heinemeier, Bennike, Krause, Margrethe Hornstrup and Thrane2008; Stiner et al. Reference Stiner, Kuhn, Weiner and Bar-Yosef1995). It is calculated from the sum of the absorption bands at 603 cm–1 and 565 cm–1 divided by the distance from the baseline to the valley between them (CI = B605 +B565)/V590) (Weiner and Bar-Yosef Reference Weiner and Bar-Yosef1990). The carbonate: phosphate ratio (C/P = B1415/B1035) of modern bone is approximately 0.23 (Garvie-Lok et al. Reference Garvie-Lok, Varney and Katzenberg2004) and decreases when calcined. Currently, the facility dates samples with an IRSF above 5 and C/P below 0.2 as calcined bone. However, it is imperative that these values are published alongside the radiocarbon dates as it is possible that their interpretation will change in the future, with the caveat that these indicies are not always comparable between laboratories, or between FTIR methods.
No method is yet available to assess whether cremated bone is affected by exogenous contamination. Although Rose et al. (Reference Agerskov Rose, Meadows, Palstra, Hamann, Boudin and Huels2019) suggest that contamination is minimal when just 5% of the bone is removed during pretreatment and Zazzo et al. (Reference Zazzo, Lebon, Chiotti, Comby, Delqué-Količ, Nespoulet and Reich2013) have accurately dated Pleistocene-aged calcined bone, van Strydonck et al. (Reference Van Strydonck, Boudin and De Mulder2009) have found some cremated bones provide erroneously young ages, suggesting that contamination may occur. To our knowledge, no work has yet assessed the preservation of calcined bone within tropical environments and dates on calcined antler (IRSF 6.9) from Con Co Ngur, Vietnam, produced an age that was younger than its context by more than 500 calibrated years (Oxenham et al. Reference Oxenham, Trinh, Willis, Jones, Domett, Castillo, Wood, Bellwood, Tromp, Kells, Piper, Pham, Matsumura and Buckley2018).
Tooth Enamel
Chemical pretreatment of tooth enamel is currently in development and is not yet able to always produce accurate radiocarbon ages, particularly beyond the late Holocene. However, it is offered by the facility as a method of producing direct minimum age estimates on skeletal remains that are not possible to date in any other way and/or where a specific chronological question is posed (e.g., whether a burial may be intrusive even though no grave cut is visible).
Where a tooth is large enough, enamel is currently dated with two methods (Wood et al. Reference Wood, Duval, Mai Huong, Tuan, Bacon, Demeter, Duringer, Oxenham and Piper2016a), and the age difference used as an indicator for degree of alteration. The surface and dentine is removed with a drill, and the enamel fractured along all existing cracks to allow soft material in cracks to be removed with the drill. After inspection under a binocular microscope to ensure all dentine and visibly recrystallized areas are removed, dust is removed by ultrasonicating in ultrapure water. A sample of at least 200 mg is crushed in a pestle and mortar under ultrapure water. Samples are ground for at least 15 minutes to ensure no large pieces remain. The sample is then split, and an aliquot of at least 120 mg is mechanically ground in a McCrone™ microniser, using agate beads, for 30 minutes in 5-minute intervals separated by 2-minute rests to ensure the sample does not heat up. The handground and micronized samples are then reacted with 1M acetic acid (1 mL/50 mg enamel) for 20 hours in a Vacutainer™. This is evacuated to <1×10–2 Torr before the addition of acid, and again after 1 hour of reaction. The cleaned enamel is rinsed 5 times with ultrapure water, freeze-dried and stored under vacuum until graphitization.
This process removes around 30% of the enamel weight. The microniser is gentler than ball mills, and preferentially fractures the enamel along grain boundaries, where substantial amounts of carbonate contaminants may be located. This means that while the acid primarily attacks the prism surfaces of the handground enamel, it attacks the crystallite surfaces (and crystallite core) in the micronized sample, often leading to an increase in age.
As with cremated bone, it is not yet possible to identify exogenous carbonates within tooth enamel. >0.8% C and unexpected δ13C values may indicate severe contamination (Wood et al. Reference Wood, Fleury, Fallon, Nguyen and Nguyen2021), and so IRMS is undertaken on the cleaned enamel. FTIR, a technique routinely used to assess preservation in the fields of δ13C and δ18O (Miller et al. Reference Ventresca Miller, Fernandes, Janzen, Nayak, Swift, Zech, Boivin and Roberts2018), is also undertaken although it is not yet possible to identify which indicies provide the best indication of exogenous carbonate contamination. Calcite has never been observed in the tooth enamel dated, and brushite formed during acidification is rare.
Keratin
A range of keratin samples are dated, including wool, nail, hair and horn. 5–20 mg is physically cleaned. The surface is drilled from horn, and a sample taken with the drill. Clean strands of hair and sections of woolen textile are picked under a binocular microscope. Both dirt (which can look like a stain) and tissue need to be separated from nails, and are easier to distinguish after soaking the nail in ultrapure water (overnight, 5°C). Using tweezers and a curved scalpel, dirt, tissue, and a thin surface layer are removed from both surfaces of the nail under a binocular microscope. The nail is kept wet by repeatedly dipping in ultrapure water. Where possible, the laboratory favors radiocarbon dating of finger nails to reduce potential contamination from hair dye and cleaning products (de la Torre et al. Reference De la Torre, Ormsby, Fuller and Santos2014).
Pretreatment was modified from O’Connell and Hedges (Reference O’Connell and Hedges1999) and Santos et al. (Reference Santos, De La Torre, Boudin, Bonafini and Saverwyns2015) and adopted in 2020. Forensic samples of hair, nail and horn are de-fatted in a mixture of dichloromethane and methanol (2:1) (2 hours, room temperature), replacing the solution after 1 hour and ultrasonicating for 15 minutes (hair) or 30 minutes (nail). After rinsing in ultrapure water, samples are subjected to an ABA pretreatment involving 0.1M HCl (30 minutes), 0.1M NaOH (1 hour) and 0.1M HCl (30 minutes) at room temperature, rinsing with ultrapure water after each treatment. Ancient samples are treated in a similar way, but without the de-fatting step. If hair is visibly dirty, the NaOH treatment and following water rinses are undertaken in an ultrasonication bath as the alkali opens the cuticle scales allowing trapped particulates to be freed more readily.
Prior to 2020, forensic keratin samples were de-fatted in the same way as forensic bone and treated with an ABA protocol consisting of 1M HCl (30 min), 0.1M NaOH (1 hour, replaced until colorless), and 1M HCl (30 min), heating to 70ºC where possible and rinsing three times in ultrapure water after each treatment.
Other Protein
Other proteinaceous samples are treated on a case-by-case basis. For example, primarily collagenous lungfish scales taken from living or recently deceased animals are not pretreated (e.g. Fallon et al. Reference Fallon, McDougall, Espinoza, Roberts, Brooks and Kind2016), and where a small amount of carbonate is present, e.g. in coral tissue, a dilute acid (e.g. 0.5M HCl, overnight, 5ºC) is used (e.g. Komugabe-Dixson et al. Reference Komugabe-Dixson, Fallon, Eggins and Thresher2016).
CARBONATE: SHELL, FORAMS, AND CORAL
The dirty and degraded surface of shell and coral, as well as other carbonates such as otoliths and celtis seeds, is removed with a drill until a dense and translucent material is left. At times this will involve removing more than half the thickness of a shell. Where an aragonite nacre is present, the nacre is separated and lightly cleaned with a drill. 5–10 mg shell is immersed in sufficient 0.1M HCl (80ºC) to remove 10–50% weight, rinsed in ultrapure water and dried in a hotblock. The sample is not crushed so that the surface is preferentially leached. Foraminifera and ostracods are picked and cleaned of sediment by the sample submitter and leached in 0.1 M HCl to remove 10–20% weight. Modern (post-1950) coral is not pretreated.
Preservation of carbonate is assessed using a combination of visual observation, Feigl’s stain and powder x-ray diffraction (XRD). In many, though not all (Webb et al. Reference Webb, Price, Northdurft, Deer and Rintoul2007), cases carbonate is recrystallized as calcite. Therefore, for calcite samples such as oyster shell, visual inspection is the only indicator of preservation and shells are not dated where there is any doubt about sample integrity. Prior to 2021, visibly degraded shells would occasionally be dated and a warning issued to the sample submitter. Since 2021, samples have not been dated where dense and translucent material with a uniform pearlescence cannot be found, or in the case of oyster or nacre, when the layers are not easily separable.
If the sample is aragonite, carbonate is screened with Feigl’s stain (Feigl Reference Feigl1937), and if >30 ka, XRD. Feigl solution is prepared by dissolving 7.2 g MnSO4.H2O per 100 mL boiling ultrapure water and adding 1g AgSO4. After cooling, the solution is passed through a baked glass fiber filter, and enough water is added to return the solution to 100 mL. 1 to 2 drops of 10% NaOH are added while stirring. After 1–2 hours, the solution is filtered a second time to remove a black precipitate, and the solution is stored in a dark glass bottle. The dry acid leached shell is covered in Feigl solution at room temperature, and after 10–20 minutes the shell is rinsed in ultrapure water and inspected under a binocular microscope. Pieces entirely covered in a black stain are selected for dating, and a scalpel is used to remove as much of the stain as possible. Where results of staining are inconclusive, results are interpreted in light of visual inspection. The texture of the shell appears crucial to the success of the staining procedure, and we have noticed the Feigl staining of nacre in particular is unreliable, perhaps due to high concentrations of protein between the carbonate crystals (Jacob et al. Reference Jacob, Soldati, Wirth, Huth, Wehrmeister and Hofmeister2008).
XRD is undertaken on aragonite samples that are > 30 ka. Before 2018, carbonate was ground under acetone, dried and passed through a 0.11 mm mesh sieve onto a quartz low background holder. XRD was undertaken using a Siemens D501 X-ray diffractometer operating at 40kV and 40mA using Cu Kα radiation, and a step size of 0.05o and speed of 0.3 minutes per degree between 25–50o 2θ. Siroquant V3 was used to quantify the calcite content with a detection limit of around 0.3wt.% calcite for a 50 mg sample. Since 2018, the sample has been hand ground in acetone, and suspended on a quartz holder and dried. XRD is carried out with a Malvern Panalytical Empyrean Series 3 which is equipped with Bragg-Brentano divergent beam optic (BBHD) and a PIXcel3D detector (1D scanning mode, 3.347° active length), using CoKα radiation. Samples are analyzed over a range of 20–58° 2 θ, with step width of 0.0131303° 2θ and a total dwell time of 200 s/step. Phase identification is carried out with the software DiffracPlus Eva 10 and the ICDD PDF-2 database and phase quantification is performed with HighScore Plus 4.8. The detection limit is about 0.1wt.% calcite.
Dissolved Inorganic Carbon
Dissolved inorganic carbon (DIC) from water is dated using a method adapted from Bragança et al. (Reference Bragança, Oliveira, Macario, Nunes, Muniz, Lamego, Abril, Nepomuceno, Solís and Rodríguez-Ceja2021). A range of waters has been dated, including both seawater and groundwater. Approximately 50 mL of water is loaded into 100 mL glass containers with crimp style septa seals in a glove bag under a N2 or He environment. The samples are then acidified with 0.5 mL 85% H3PO4 with a Hamilton Gastight™ syringe and needle (Terumo™ 0.45×13 mm) for 24 hours at room temperature to react. CO2 is cryogenically collected over 10 minutes with water removed using dry ice and ethanol. A second 50 mL aliquot is prepared if carbon content is low. Prior to this we precipitated SrCO3 from groundwater DIC. In brief, we added 1–2 g of SrCl2 per L of groundwater. Several pellets of NaOH were used to increase pH and speed up the precipitation. The sample was centrifuged, water removed and SrCO3 was rinsed 3–5 times with ultrapure water, centrifuging between each rinse, before freeze-drying.
Combustion, Acidification, and Graphitization
Organic samples, normally containing 0.25–1 mg C, are weighed into an Ag cup (Sercon™ SC1081) and placed into a quartz tube (Infusil™ silicaware 4×6×200 mm) with 30 mg CuO (Sercon SC0003). An Ag cup is used rather than the typical Ag wire to avoid powdered samples or collagen from adhering to the quartz tube wall and hindering sealing. Before use, Ag and CuO are ultrasonicated in methanol (repeated until no dust is released), and baked (900°C, 3 hours), and quartz tubes are baked (900ºC, 3 hours). Samples are evacuated to < 3×10–3 Torr and sealed prior to combustion in a muffle furnace (900ºC, 6 hours). CuO is the major source of carbon contamination in the combustion/graphitization process, and in addition to testing each batch for both young and old carbon, efforts have been made to reduce the quantity of CuO. Prior to 29/10/2019, 60 mg CuO was used. This was reduced to 20 mg (29/10/2019–12/12/2019). While sufficient to combust the samples, samples and standards sometimes failed to graphitize presumably because gaseous contaminants such as SO2 were not removed with the reduced Cu. 30 mg CuO appears sufficient for the majority of samples.
Carbonates, including SrCO3 from DIC, tooth enamel and cremated bone are placed in a 10 mL Vacutainer™ vial and evacuated to <3×10–3 Torr. 0.3–0.4 mL 85% H3PO4 is added to carbonates with a Hamilton GastightTM syringe and needle (TerumoTM 0.45×13 mm). 1 mL is used for enamel and cremated bone. The reaction is left at at 80ºC until complete.
CO2 is cryogenically collected and purified on a vacuum line built with Swagelock™ Ultratorr, using a dry ice-ethanol trap. CO2 is collected in a reaction rig fitted with KimaxTM culture tubes (6×50 mm) for graphitization with ultra high purity H2 over an Fe catalyst (560ºC, 6 hours or until reaction is complete). Fe is produced from 3.0–3.3 mg Fe2O3 (Aldrich™ 529311) by reaction with H2 (1000 Torr, 400ºC, 90 minutes). Granular magnesium perchlorate (Alfa Aesar™ 11626) is used to remove water generated during the graphitization process (Santos et al. Reference Santos, Southon, Druffel-Rodriguez, Griffin and Mazon2004).
Our standard reactors have a volume of ca. 3.6 cm3 and are used to produce samples between 0.25–1 mg C. Samples between 0.15 mg–0.25 mg C are dated using a small reaction vessel, and matched in size to primary and secondary standards. The small reaction vessels were machined in the Research School of Earth Sciences mechanical workshop and are combined with shortened, pre-baked borosilicate tubes to hold Mg(ClO4)2. These modifications result in decreased the reactor volume to ∼2.1cm3. Samples below 0.15 mg are only dated in specific cases, for example, where the sample is very young and larger samples cannot be recovered. In these cases graphitization temperature is reduced to 450ºC (Strzepek Reference Strzepek2014).
Samples containing large amounts of gaseous contaminants, such as sediments, cremated bone and tooth enamel, require an extra cleaning step to remove SO2. These samples are converted to CO2, either by acidification with phosphoric acid (cremated bone and enamel) or combustion (organics), as described above. The CO2 is cryogenically purified as described for graphitization but is collected in a quartz tube containing CuO wire and Ag foil. This is sealed and heated to 900ºC for 6 hours, and the cleaned CO2 is then cryogenically collected and purified for a second time, before graphitization. This extra step adds a small amount of laboratory contamination, and an extra background correction has been calculated (Wood et al. Reference Wood, Fleury, Fallon, Nguyen and Nguyen2021).
Data Quality: Standards and Duplicates
Approximately 1 in 20 samples is duplicated from sampling to AMS measurement, to assess whether quoted errors are appropriate. A range of pretreatment-specific standards are processed (Table 2). Where possible, samples are matched to the material and age of the unknowns. Where no standard is available, coal for organics or marble for inorganics is normally treated and dated. Data reproducibility is explored in Fallon et al. (Reference Fallon, Wood and Esmayin prep).
Table 2 Secondary pretreatment standards routinely used in the ANU radiocarbon facility.

ACKNOWLEDGMENTS
RW was supported by an Australian Research Council DECRA fellowship (DE150100070) for development of the enamel protocol, and DP160100811 supported the introduction of the cremated bone protocol. Those supplying materials for standards are acknowledged in Table 2. Ulrike Troitzsch is thanked for XRD analysis, Fiona Petchey and David Chivall are thanked for discussions about Feigl’s solution, and Penny King is thanked for advice and help with FTIR. Past students and staff of the lab are thanked for their input into the development, refinement and use of laboratory protocols, including Susan Alford, Kelly Strzepek, Aimée Komugabe, Yang Wu, Bowen Fang, Graham Nash, Andre Barros Fleury, and Stephanie August.






 Open access
Open access