


Consumption of refined sugars has markedly increased over the past decades(Reference Wolf, Bray and Popkin1). In the Western world, these are mostly consumed under two forms: either as sucrose, mainly extracted from beet, and constituted of one molecule of fructose linked to one molecule of glucose; or as high-fructose corn syrup, which consists in a mixture of free fructose and glucose, the most common form being characterised by a fructose:glucose ratio of 55:45(Reference Hanover and White2). Recently, the drastic increase in high-fructose corn syrup consumption at the detriment of sucrose has raised much concern(Reference Bray, Nielsen and Popkin3). Several authors have suggested that such increase in free fructose consumption may be linked to the development of obesity and the metabolic syndrome. Indeed, both in rodents and human subjects, high-fructose diets (HFrD) lead to hypertriacylglycerolaemia, insulin resistance and accumulation of ectopic lipid in the liver and the muscle, known as intrahepatocellular lipids (IHCL) and intramyocellular lipids (IMCL), respectively(Reference Havel4–Reference Lê and Tappy6). These deleterious effects were attributed to the fact that fructose, by bypassing the major regulatory point of glycolysis, rapidly leads to an excess of triose phosphates in hepatocytes, which may be used as substrates for de novo lipogenesis. Several rodents(Reference Carmona and Freedland7, Reference Clark, Rognstad and Katz8) and human studies(Reference Parks, Skokan and Timlin9) have previously shown that fructose was a more potent stimulator of lipogenesis than glucose. However, most of these studies were performed in an acute setting, and it remains therefore unknown whether chronic fructose-induced alterations of lipid homoeostasis are due to specific fructose properties, or are merely the result of energy and/or sugar overloading. The aim of the present study was to compare the effects of a hypercaloric 7-d HFrD v. high-glucose diet (HGlcD) on ectopic lipids, glucose homoeostasis and plasma lipid profile.
Subjects and methods
Subjects
Eleven healthy non-smoking male volunteers (24·6 (sem 0·6) years; means with their standard errors) participated in the study. According to a physical examination and a brief medical history, all subjects were in good health with a BMI between 19 and 25 kg/m2 and were moderately physically active ( < 1 h/week). They were not taking any medications and did not regularly consume alcohol or sugar-sweetened beverages.
Study design and diet
Each subject consumed, in a crossover randomised order, the following diets: (1) a 7-d weight maintenance diet (total energy intake equal to predicted basal energy requirement(Reference Owen10) × 1·6), containing 55 % carbohydrate (of which 11 % simple sugars), 30 % fat and 15 % protein; (2) the same weight maintenance diet supplemented with 3·5 g fructose/kg fat-free mass per d: HFrD; or (3) the weight maintenance diet supplemented with 3·5 g glucose/kg fat-free mass per d: HGlcD. Both HFrD and HGlcD corresponded to an energy overload corresponding to +35 % energy requirements. The study was performed on an outpatient basis, and during the 3 d preceding the metabolic investigations, subjects were provided with all the dietary constituents as pre-packed food items with instruction as to how and when to consume them. Fructose and glucose were administered as a 20 % solution with the three main meals. A 2–3-week washout period separated the three dietary conditions. Leisure sport activity was restricted to < 1 h/week throughout the study period. Compliance was assessed by interview.
The present study was conducted according to the guidelines laid down in the Declaration of Helsinki, and all procedures involving human subjects/patients were approved by the ethical board of Lausanne University School of Biology and Medicine. Written informed consent was obtained from all subjects.
Metabolic investigation
Subjects reported at 07.00 hours to the metabolic unit of the Lausanne University Hospital after a 10-h fast. Upon arrival, subjects were asked to void, and body composition was estimated from subcutaneous skinfold thickness measurements at the biceps, triceps, subscapular and suprailiac sites(Reference Durnin and Womersley11). Participants thereafter rested quietly in a bed in a semi-recumbent position, and an indwelling catheter was inserted into the vein of the right wrist for blood sampling. A second indwelling catheter was inserted into an antecubital vein of the other arm for infusion of 6,6-2H2 glucose. Fasting hepatic glucose output was assessed in basal condition after a 2-h 6,6-2H2 glucose infusion (bolus: 2 mg/kg; continuous: 20 μg/kg per min) by glucose isotope dilution analysis using Steele's equations for steady-state conditions(Reference DeBodo, Steele and Altszuler12). Blood was collected during baseline for measurement of plasma concentrations of glucose, lactate, insulin, non-esterified fatty acids, β-hydroxybutyric acid, uric acid, total TAG, as well as VLDL, LDL and HDL subfractions, alanine aminotransferase and leptin. Energy expenditure and substrate utilisation were continuously measured by indirect calorimetry (ventilated canopy) from 08.00 to 10.00 hours using the equations of Livesey & Elia(Reference Livesey and Elia13).
Fasting hepatic insulin sensitivity index was calculated as(Reference Matsuda and DeFronzo14):
Analytical procedures
Plasma was immediately separated from blood by centrifugation at 4°C for 10 min at 3600 rpm and stored at − 20°C. Colorimetric methods were used to assess plasma concentrations of NEFA (kit from Wako Chemicals, Freiburg, Germany) and TAG (kit from Biomérieux Vitek, Inc., Durham, Switzerland). Commercial RIA kits were used for the determination of plasma insulin and leptin (LINCO Research, St Charles, MO, USA). Subfractions of lipoproteins were separated by ultracentrifugation. β-Hydroxybutyric acid and lactate concentrations were determined enzymatically using kits from Boehringer (Mannheim, Germany). Plasma glucose concentration was measured by the glucose oxidase method using a Beckman glucose analyzer II (Beckman Instruments, Fullerton, CA, USA). Plasma 6,6-2H2 glucose isotopic enrichment was measured by GC–MS (Hewlett Packard Instruments, Palo Alto, CA, USA), as previously described(Reference Schneiter, Gillet and Chioléro15).
1H Magnetic resonance spectroscopy
All 1H-magnetic resonance spectroscopy examinations were performed on a clinical 1·5 T MR scanner with data acquisition (single-voxel localisation with 20 ms echo time) and processing similar to a protocol described earlier for IMCL(Reference Lê, Faeh and Stettler16) and IHCL(Reference Bortolotti, Kreis and Debard17). Fat content was expressed in mmol/kg.
Statistical analysis
Data are expressed as means with their standard errors. Statistical analyses were performed with STATA version 8.2 and P < 0·05 was considered statistically significant. Because IHCL values were not normally distributed, they were converted into log values before statistical analysis. All the data were analysed by using repeated-measures ANOVA. Post hoc comparisons were done by using the Student's paired t test.
Results
All subjects significantly gained weight after both hypercaloric diets (Table 1). HFrD increased VLDL by 59 (sem 24) % (range − 28 to +197), P < 0·05 and IHCL by 52 (sem 13) % (range − 27 to +203), P < 0·05. With HGlcD, increases in similar magnitudes were observed, but did not reach statistical significance due to large interindividual variations in the response to hypercaloric diets (VLDL-TAG: +31 (sem 20) % (range − 44 to +139), P = 0·11; IHCL: +58 (sem 23) % (range − 29 to +226), P = 0·06 (Fig. 1). IHCL and VLDL-TAG were not different with HFrD and HGlcD. No change was observed in fasting glycaemia, insulin and alanine aminotransferase concentrations (Table 1). Both diets significantly decreased NEFA and ketone bodies (Table 1). IMCL significantly increased only after the HGlcD (fructose: +49 (sem 23) % (range − 23 to +239), NS; glucose: +84 (sem 86) % (range − 20 to +319), P < 0·05). Carbohydrate oxidation increased with a concomitant decrease in lipid oxidation after both hypercaloric diets. Hepatic glucose output increased after both diets (HFrD: +5 %, P < 0·05; HGlcD: +5 %, P = 0·05; Table 1). Hepatic insulin sensitivity index decreased to the same extent after HFrD and HGlcD, but failed to reach significance.
Table 1 Anthropometric and metabolic parameters after the weight maintenance, the high-fructose (HFrD) and high-glucose diets (HGlcD)
(Mean values with their standard errors)

ALAT, Ala aminotransferase; ASAT, aspartate aminotransferase; HGO, hepatic glucose output.
* HFrD v. weight maintenance.
† HGlcD v. weight maintenance.
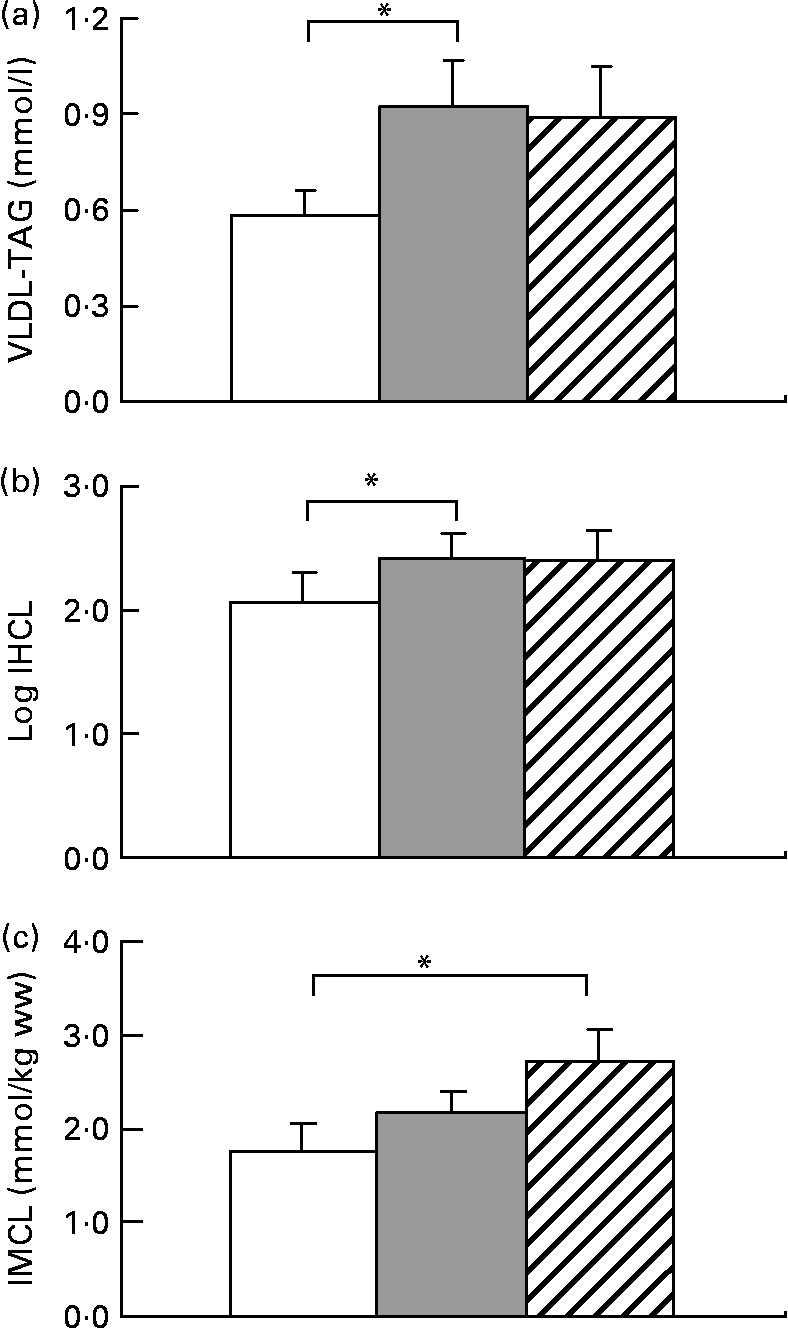
Fig. 1 Data are expressed as means with their standard errors; n 11; VLDL-TAG (a), intrahepatocellular lipids (IHCL) (b) and intramyocellular lipids (IMCL) (c). * Mean values were significantly different v. weight maintenance diet (P < 0·05) by Student's t test. ww, Wet weight. □, Isocaloric; ![]() , high fructose; ▨, high glucose.
, high fructose; ▨, high glucose.
Discussion
It has been known for decades that a HFrD leads, in human subjects or in rodents, to several features of the metabolic syndrome, including hypertriacylglycerolaemia(Reference Lê, Faeh and Stettler16, Reference Bantle, Laine and Thomas18, Reference Chong, Fielding and Frayn19), insulin resistance(Reference Pagliassotti and Prach20–Reference Faeh, Minehira and Schwarz22) and ectopic lipid deposition(Reference Pagliassotti, Prach and Koppenhafer21). However, in several of these studies, high fructose intake was also associated with a high energy intake, but was not compared to a high glucose intake. This made it impossible to sort out the effects of fructose per se and those of overfeeding with simple sugars. In the present study, we show that a hypercaloric HGlcD leads to many of the metabolic alterations associated with HFrD.
Few studies have directly compared the effects of fructose v. glucose. When administered as part of a test meal, fructose stimulates hepatic de novo lipogenesis to a greater extent than glucose(Reference Parks, Skokan and Timlin9), and causes higher postprandial and 24-h triglyceridaemia(Reference Teff, Elliott and Tschöp23, Reference Stanhope, Griffen and Bair24). These differences observed after acute administration are best explained by the distinct metabolism of these two carbohydrates. Unlike glucose, fructose metabolism does not require the action of insulin. After ingestion, fructose is directly delivered in the liver, which constitutes the main site of its metabolism. In hepatocytes, fructose is degraded into triose phosphates, which can be diverted into one of the following pathways: oxidation, lactate and glucose production or de novo lipogenesis(Reference Mayes25).
The chronic effects of a HFrD v. a HGlcD have been less described. In a previous study, Bantle et al. (Reference Bantle, Raatz and Thomas26) found that fructose, but not glucose, increased fasting and postprandial TAG in healthy men. However, both sugars were consumed as part of an isocaloric, weight maintenance diet. In obese human subjects, overfeeding with fructose, but not with glucose, has been reported to produce a slight increase in intra-visceral fat over a 10-week period and to enhance postprandial plasma TAG(Reference Stanhope, Schwarz and Keim27). In rodents, hypercaloric HFrD, but not HGlcD, led to higher IHCL deposition and plasma TAG after 2 weeks(Reference Roglans, Vila and Farre28). Analysis of liver tissue revealed that both fructose and glucose consumption stimulated lipogenic genes expression; however, only fructose decreased hepatic expression and activity of genes involved in lipid oxidation. It was therefore suggested that fructose-induced inhibition of hepatic lipid oxidation may be responsible for the IHCL accumulation(Reference Roglans, Vila and Farre28).
The present results are at odds with some of these reports. Fructose overfeeding indeed increased IHCL and VLDL-TAG, as reported in rodents(Reference Roglans, Vila and Farre28) and in human subjects(Reference Le, Ith and Kreis29). The presently reported increase in IHCL was, however, of a smaller magnitude than in our previous study(Reference Le, Ith and Kreis29), and showed considerable inter-individual variability. Part of this variability may be explained by genetic factors, since we have reported that offsprings of patients with type 2 diabetes have fructose-induced increases in IHCL(Reference Le, Ith and Kreis29). Inter-individual differences in insulin sensitivity may also play a role since hepatic insulin resistance has been shown to be strongly associated with intrahepatic fat(Reference Kotronen, Juurinen and Tiikkainen30). However, and in contrast with what was reported in rodents(Reference Roglans, Vila and Farre28), we observed that glucose overfeeding during 7 d also increased IHCL and plasma VLDL-TAG. When expressed as the percentage change from values observed with the control, weight maintenance diet, fructose and glucose led to similar increases in IHCL and plasma VLDL-TAG. However, interindividual variations were more important with glucose than with fructose. Due to this variability, the increases in IHCL and VLDL-TAG observed after glucose fell short of reaching statistical significance. This more inconstant increase in IHCL with glucose compared to fructose may be possibly due to the fact that the pathways used for hepatic lipid deposition may differ between these two sugars. Fructose is essentially metabolised in liver cells, and stimulation of hepatic de novo lipogenesis may be directly linked to intrahepatic lipid storage and VLDL-TAG secretion(Reference Faeh, Minehira and Schwarz22), while only part of the glucose administered is metabolised in liver cells; hepatic lipid storage and dyslipidaemia during glucose overfeeding are therefore likely to rely mainly on intrahepatic re-esterification of NEFA.
Both glucose and fructose overfeeding led to subtle, but significant changes in fasting hepatic glucose metabolism, and tended to decrease hepatic insulin sensitivity index. Although the mechanisms responsible for this slight decrease in hepatic insulin sensitivity remain unknown, it is tempting to speculate that it is linked with intrahepatic fat deposition.
Unexpectedly, glucose overfeeding led to a substantial and significant increase in IMCL deposition, while the increase observed with fructose was of smaller magnitude and did not reach statistical significance in this group of healthy subjects. We have, however, reported previously that fructose overfeeding also increases IMCL in healthy subjects and in offsprings of patients with type 2 diabetes, and here again, a considerable inter-individual variation is likely to be responsible for failure to reach statistical significance in the present study. The apparent larger increase in IMCL after glucose may nonetheless reflect the different pathways used for the metabolism of these two sugars. Contrarily to fructose, a major portion of a glucose load is directly metabolised in muscle, where it stimulates glucose oxidation(Reference Kelley, Mitrakou and Marsh31). A concomitant inhibition of muscle lipid oxidation may therefore favour deposition of intramyocellular lipid.
Glucose and fructose overfeeding led to similar decreases in plasma NEFA, indicating suppression of adipose tissue lipolysis. This observation may appear surprising since insulin is the major factor inhibiting lipolysis and glucose is expected to produce much larger increase in insulin concentrations. Adipocytes are, however, extremely sensitive to insulin, and the slight increase in plasma insulin elicited by oral fructose has been reported to be sufficient to significantly inhibit lipolysis(Reference Tappy, Randin and Felber32). Fructose overfeeding also suppressed basal, post-absorptive NEFA, as previously reported(Reference Abdel-Sayed, Binnert and Le33). The mechanisms remain hypothetical at this point, but may involve changes in the expression of genes involved in lipid storage and/or lipid oxidation.
In summary, our present data indicate that a short-term overfeeding with either fructose or glucose leads within 7 d to several potentially deleterious metabolic alterations in healthy human subjects. Both sugars increased plasma TAG, which may increase cardiovascular risk(Reference Jonkers, Smelt and van der Laarse34). Both sugars also led to intrahepatic fat deposition, and such effect may, in the long term, favour the development of non-alcoholic fatty liver disease. They also led to slight alterations of hepatic insulin sensitivity index. Finally, glucose overfeeding led to a significant deposition of ectopic fat in muscle, which may, in the long term, favour muscle insulin resistance(Reference Kelley and Goodpaster35). Both diets were hypercaloric, and it is possible that excess energy intake rather than specific effects of sugars was responsible for these metabolic alterations. In support of this hypothesis, we have recently reported that a short-term overfeeding with 30 % excess energy as saturated fat also increased IHCL concentrations(Reference Bortolotti, Kreis and Debard17). Fat overfeeding, however, failed to increase plasma TAG concentrations, suggesting that sugars may have additional effects on plasma lipid concentrations. From a practical point of view, these results suggest that high energy and high sugar intakes may confer a risk for the development of metabolic disorders. In this perspective, reducing sugar intake may be a primary target for prevention of metabolic diseases.
Acknowledgements
The present work was supported by grant no. 121995 from the Swiss National foundation for Science. E. T. N. S., K.-A. L. and L. T. performed the metabolic tests; K.-A. L. and L. T. designed the experiment; M. I., R. K. and C. B. performed the NMR measurements; E. T. N. S. and K.-A. L. performed the statistical analysis; K.-A. L. drafted the manuscript; all authors revised the manuscript. The study was performed at the Department of Physiology, University of Lausanne, Lausanne, Switzerland. We thank the staff of the Department of Physiology of Lausanne for their excellent assistance, and all the volunteers for their participation and commitment. The authors state that there are no conflicts of interest.


