



Introduction
Legionella infections are an important cause of community- and hospital-acquired pneumonia, known as Legionnaires’ disease (LD). In addition, Legionella can cause water-based outbreaks of public health implications [Reference Principe, Tomao and Visca1]. Legionella infections or legionellosis varies in severity from a mild febrile illness known as pontaic fever to a serious and sometimes fatal form of atypical pneumonia called LD [Reference Mercante and Winchell2]. Legionellosis is a notifiable disease in all European Union countries [Reference Beaute3], while in developing countries it is under-reported; this is mainly due to underdiagnosis by clinicians who only rarely test for LD before implementing antibiotic therapy that is likely to cover Legionella spp [Reference Diederen4].
In the West Bank, Palestine, LD is not a notifiable disease, and prior to this study, no information about the prevalence of Legionella spp. in environmental and clinical samples was available. Legionella pneumophila is the aetiological agent of approximately 90% of legionellosis cases [Reference Yu5], and L. pneumophila serogroup 1 (sg 1) is the predominant cause [Reference Aurell6]. Isolation of L. pneumophila by culture is considered the ‘gold standard’ for the diagnosis of LD [Reference Mercante and Winchell2], but culture-confirmed diagnosis of Legionella infections is often hampered by the demanding culture requirements that need professional personnel and by early antibiotic treatment [Reference Qin7, Reference Roig and Rello8].
According to the WHO Essential Medicines and Health Products report, there is irrational use of drugs in Palestinian National Authority (PNA) health institutions, and most patients use antibiotics prior to hospital admission [Reference Obeidallah9, 10], making it difficult to isolate L. pneumophila strains from clinical specimens. On the other hand, molecular diagnostic techniques for L. pneumophila from respiratory samples, such as polymerase chain reaction (PCR), are well established and increasingly adopted worldwide [Reference Mercante and Winchell2, Reference Divan Khosroshahi, Naserpour Farivar and Johari11]. Endpoint and real-time PCR offer substantial advantages over cultivation for the detection and quantification of L. pneumophila, including high sensitivity, specificity and time efficiency [Reference Diederen12–Reference Peci, Winter and Gubbay15]. For epidemiological studies, in addition to detection and diagnosis, several rapid and discriminatory methods have been developed for Legionella typing; DNA banding pattern-based and DNA sequencing-based genotyping (among others, Ref) are widely used methods for subtyping of bacteria. Pulsed-field gel electrophoresis, an example of DNA banding method, is based on enzymatic restriction, but it requires high-quality DNA, which is poorly applicable to human or environmental samples, and may lack the resolution power to distinguish bands of nearly identical size.
Restriction fragment length polymorphism (RFLP) patterns are mainly determined through the specific combination of restriction enzymes and nucleic acid probes (Southern blotting). RFLP analysis requires large amounts of high-quality genomic DNA, which can limit its application in many cases. In addition, it is time and labour consuming, especially when coupled with Southern blotting, and involves detection systems that use either radioisotopes or complex biochemistry.
On the other hand, multiple-locus variable number tandem repeat analysis (MLVA) based on the number of tandem repeats per locus that may vary dramatically between strains within a given species. MLVA is a PCR-based genotyping method based on the polymorphic analysis of multiple variable number tandem repeat loci on the chromosome; it is a rapid, easy to perform, inexpensive and reproducible genotyping method with high resolution. Nevertheless, by comparison of genotyping methods, sequence-based typing (SBT) is considered more rapid and easier to perform and provides unambiguous results, and hence is considered the current gold standard for high-resolution genotying of L. pneumophila [Reference Luck16–Reference Scaturro19]. SBT is based on the comparison of gene sequences from seven loci (flaA, pilE, asd, mip, mompS, proA and neuA) [Reference Luck16, Reference Blyth, Adams and Chen17]. Another commonly used test for diagnosis of LD is the urinary antigen (UAG) [Reference Aurell6]. UAG is known for its high sensitivity (96%) and specificity (99.9%). However, the UAG test detects only L. pneumophila serogroup 1 (Lp1) while non-Lp1 LD is missed [Reference Chen20] and its diagnostic window is <2 weeks after infection. Still, because of its high cost, the UAG test is unlikely to be widely used in economically underdeveloped regions like the West Bank.
In the present study, our aim was to assess the prevalence of L. pneumophila in respiratory secretions from suspected pneumonia patients in the West Bank, by implementing molecular methods for the detection of L. pneumophila, and to determine the genotype of the PCR-positive samples in situ, by nested PCR sequence-based typing (NPSBT). For this purpose, we performed a 2-year prospective survey to detect L. pneumophila in respiratory tract secretions from patients with suspected pneumonia from a central hospital in the West Bank. Nested SBT was used for genotyping of the PCR-positive specimens. The need for such a study was eminent to evaluate nosocomial threats of L. pneumophila, especially that our previous work in the same laboratory showed the abundant prevalence of L. pneumophila in water systems in eight hospitals in the West Bank, Palestine (unpublished data)
Methodology
Study site and inclusion criteria
The clinical samples were collected from suspected pneumonia patients admitted to a hospital in East Jerusalem in the period between September 2014 and June 2016, after signing a written informed consent form and ethical approval from Al-Quds University and the Hospital's Ethical Committee.
Sampling of clinical and environmental material
Bronchoalveolar lavage (BAL) samples (n = 74) and sputum samples (n = 121) were collected from suspected pneumonia patients by the pulmonologist; sterile containers with screw caps were sent to Bacteriology laboratory of the Hospital. The samples were stored at 4 °C until received by the Microbiology Research Laboratory at Al-Quds University within 24 h.
Drinking water was collected from the main water source of each site and several biofilm swabs from faucets, showerheads and hoses. A total of 5 litres of each water sample was collected and filtered onto sandwich membrane filters composed of nucleopore-filter (Nuclepore Track-Etch Membrane, MB 90 mm, 0.2 µm, Whatman, England) and glass fibre-microfilter (GF/F) (GFF, 90 mm, Whatman, England). Swabs from the respective surfaces were obtained for DNA extraction using sterile cotton swabs (Cotton Tipped Applicator, China). Both types of environmental samples were store frozen (−20 °C) for later analysis.
Culture of sputum and BAL samples
BAL and sputum samples (sputum samples were treated thermally (56 °C for 10 min)) and then inoculated onto GVPC (Glycine–Vancomycin–Polymyxin Cycloheximide; Heidelberg, Germany) Legionella-selective agar medium. Plates were incubated at 37 °C for 7 days. Five colonies of each positive sample were selected and re-isolated on GVPC plates. Identification of Legionella spp. was confirmed by the ability to grow on GVPC and inability to grow on blood agar (M073, HI media, India) (L-cysteine free) plates. Positive samples were re-streaked on BCYE and further identified by an agglutination test using Legionella Latex Test (Oxoid DR0800M, England) according to manufacturer's instruction. The test allows a separate identification of L. pneumophila serogroup 1 (sg 1) and serogroups 2–14.
DNA extraction from clinical and environmental material
The BAL or sputum samples were stored at 4 °C until processed within 24–48 h; samples were vortexed for 10 s and 1–2 ml of each sample was transferred to a fresh sterile tube. Genomic DNA was extracted from each BAL and sputum sample by using DNA extraction kit (Qiagen, 69506, Germany). Briefly, 2 ml of BAL and 1 ml of sputum was centrifuged at 17 000g for 10 min then the pellet was suspended in 250 µl of sputum lysis buffer (20 mM Tris-HCl, 2 mM EDTA, 1.2% Triton X-100 (9002-93-1, Sigma-Aldrich, Germany) [pH 8.0]) containing 10 mg/ml lysozyme (62970, Fluka, Sigma-Aldrich, Switzerland) with 250 µl of molecular grade water (Promega, USA), then incubated at 37 °C for 30 min (with shaking at 1500 rpm). The samples were centrifuged at 17 000g for 15 min, and then the supernatant was discarded. Eighteen microlitres of ATL buffer was added with 20 µl of proteinase K (15 mg/ml) and incubated at 56 °C for 1 h (with shaking at 1500 rpm), then 200 µl of AL buffer was added to the sample and incubated at 70 °C for 10 min, then 200 µl of ethanol (96–100%) was added and mixed by vortexing, the samples were loaded onto the spin column and centrifuged at 17 000g for 1 min, then 500 µl of AW1 buffer was added and centrifuged at 6000g for 1 min, then 500 µl of AW2 buffer was added, then centrifuged at 17 000g for 3 min, then the spin column was placed in a new 2 ml collection tube and 100 µl of AE buffer or distilled water was added then centrifuged at 6000g. Extracted DNA was stored at −20 °C.
Biofilm swabs from anterior surfaces of faucets, showerheads or shower hoses from wards of patients positive for L. pneumophila were incubated with 220 µl lysis buffer (2x TE) containing 10 mg/ml lysozyme (Sigma) and 15 mg/ml proteinase K (Qiagen) for 20 min at 37 °C. Three hundred and fifty microlitres of AL-buffer (DNeasy kit, Qiagen) were added to two replicate swabs followed by incubation for 5 min at 70 °C. The lysate was removed from the swab by a short spin down and absolute ethanol was added to the lysate (ratio lysate/ethanol 2:1). The mixture was applied to the spin-column of the kit, then the manufacturer's instructions were followed. Nucleic acids were eluted from the columns with DNase/RNase-free water and stored at −20 °C. More details on the sampling and DNA extraction of water and biofilm samples are provided by Henne et al. [Reference Henne21].
Screening of BAL and sputum samples for L. pneumophila by PCR
Endpoint PCR assays targeting the 16S rRNA gene were used for identification of L. pneumophila in BAL and sputum samples. Primers for L. pneumophila species, L1F 5′-CCTGGGCTAACCTGGGAC-3′ and L1R 5′-CTTAGACTCCCCACCATCACAT-3′, were purchased from Hylabs (Rehovot, Israel). AccuPower® HotStart PCR PreMix was used for PCR amplification; to each AccuPower® HotStart PCR PreMix tube, a mixture of 0.8 µl (10 mmol) forward primer (L1F), 0.8 µl (10 mmol) reverse primer (L1R) and 4 µl (100 ng/μl) DNA template was distributed into each PreMix, and 14.4 µl molecular grade water (Promega) was added. PCR amplification was done using a thermal cycler (model 1861096, Biorad, USA) at the following conditions: denaturation at 94 °C for 5 min followed by 35 cycles of denaturation at 94 °C for 45 s, annealing at 55 °C for 60 s, elongation at 72 °C for 45 s and final elongation at 72 °C for 5 min. PCR products were analysed by agarose gel electrophoresis to check for the correct amplicon size (544 bp). DNA of Legionella reference strain L. pneumophila subsp. pneumophila Philadelphia 1 ATCC33152 was used as positive DNA control throughout the study. Molecular grade water (Promega) was used as negative control in all PCR assays.
Molecular typing by NPSBT
NPSBT was used to obtain typing data for L. pneumophila from BAL and sputum samples as well as from environmental samples (biofilm) collected from different wards of the same hospital. The starting material used for genotyping was DNA of samples which were confirmed to be positive for the presence of L. pneumophila by PCR. The procedure of the European Working Group for Legionella Infections (EWGLI), Nested-SBT (version 2.0) was applied as detailed at http://www.hpa-bioinformatics.org.uk/legionella/legionella_sbt/php/sbt_homepage.php.
In brief, the protocol included two rounds of PCR: the first round of PCR was carried out with seven primer pairs (flaA, pilE, asd, mip, mompS, proA, neuA) [Reference Gaia22], which bind externally to those used in the second-round PCR for another seven primer pairs. PCR products of the first round was the starting material for the second-round PCR; the PCR products from the second round were sent to Hylabs, Israel, for purification and sequencing; the second-round primers were used as sequencing primers. Sequencing results were submitted to the online Legionella SBT Quality Tool www.hpa-bioinformatics.org.uk/cgibin/legionella/sbt/seq_assemble_legionella1.cgi which was used to assign individual allele numbers based on comparison of the sequences to STs of L. pneumophila in the database. For each isolate, the combination of seven alleles was defined as a seven-digit allelic profile by using the predetermined order flaA, pilE, asd, mip, mompS, proA and neuA (e.g. 1-4-3-1-1-1-1) and a ST represented by a number (e.g. ST 1); if less than seven-allele profile was obtained (5–6), it was usually sufficient to identify the strain as belonging to one or two STs, three- or four-allele profiles sufficient to differentiate one profile from another.
Results
Clinical data
The study included 195 respiratory specimens including 74 BAL and 121 sputum samples collected from suspected pneumonia patients that were hospitalised at a hospital in East Jerusalem, in the period between September 2014 and June 2016. Patients that had showed more than one positive result were counted as one considering the department of the first positive sample.
The age of the patients ranged between 3 months and 86 years. Patients were hospitalised in the following departments: internal medicine, intensive care unit (ICU), paediatric, paediatric ICU, surgery and gynaecology (Fig. 1).
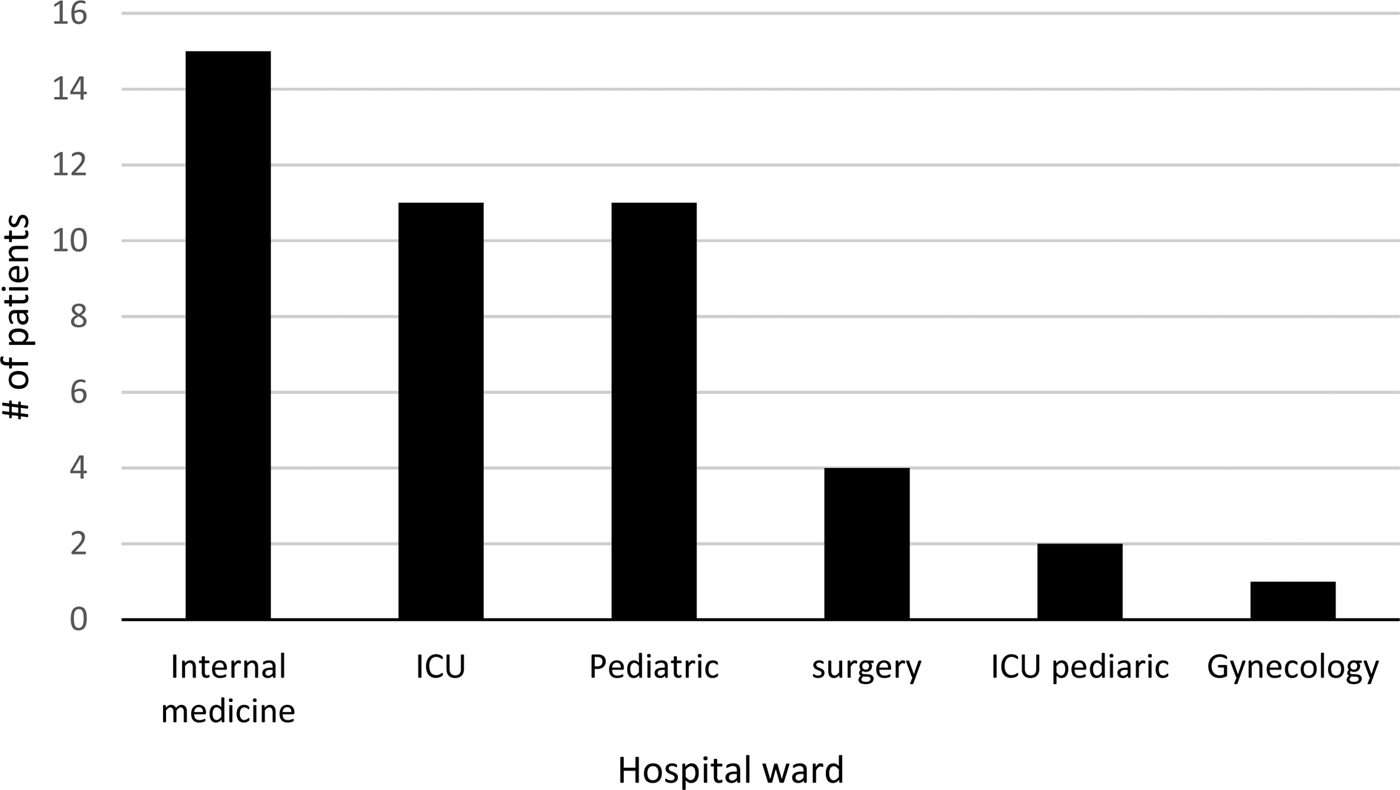
Fig. 1. Distribution of Legionella pneumophila – positive samples (n = 44) by PCR, from pneumonia patients according to hospital departments.
Detection of L. pneumophila in respiratory samples by cultivation
All respiratory samples (74 BAL and 121 sputum samples) were tested by cultivation-dependent analysis by plating on GVPC-selective media. Only one of the total 195 samples was positive for L. pneumophila by routine bacteriological culture method. This very low yield may be explained due to the regimen of antibiotics heavily used by patients prior to hospitalisation.
Prevalence of L. pneumophila in respiratory samples determined by PCR
PCR analysis of 195 respiratory samples for L. pneumophila revealed 23% positive and 77% negative samples in all analyses. This molecular detection of L. pneumophila in the sputum samples (n = 121) resulted in 18 (15%) positive and 103 (85%) negative samples, whereas analysis of the BAL samples (n = 74) showed 26 (35%) positive and 48 (65%) negative samples (Fig. 2).
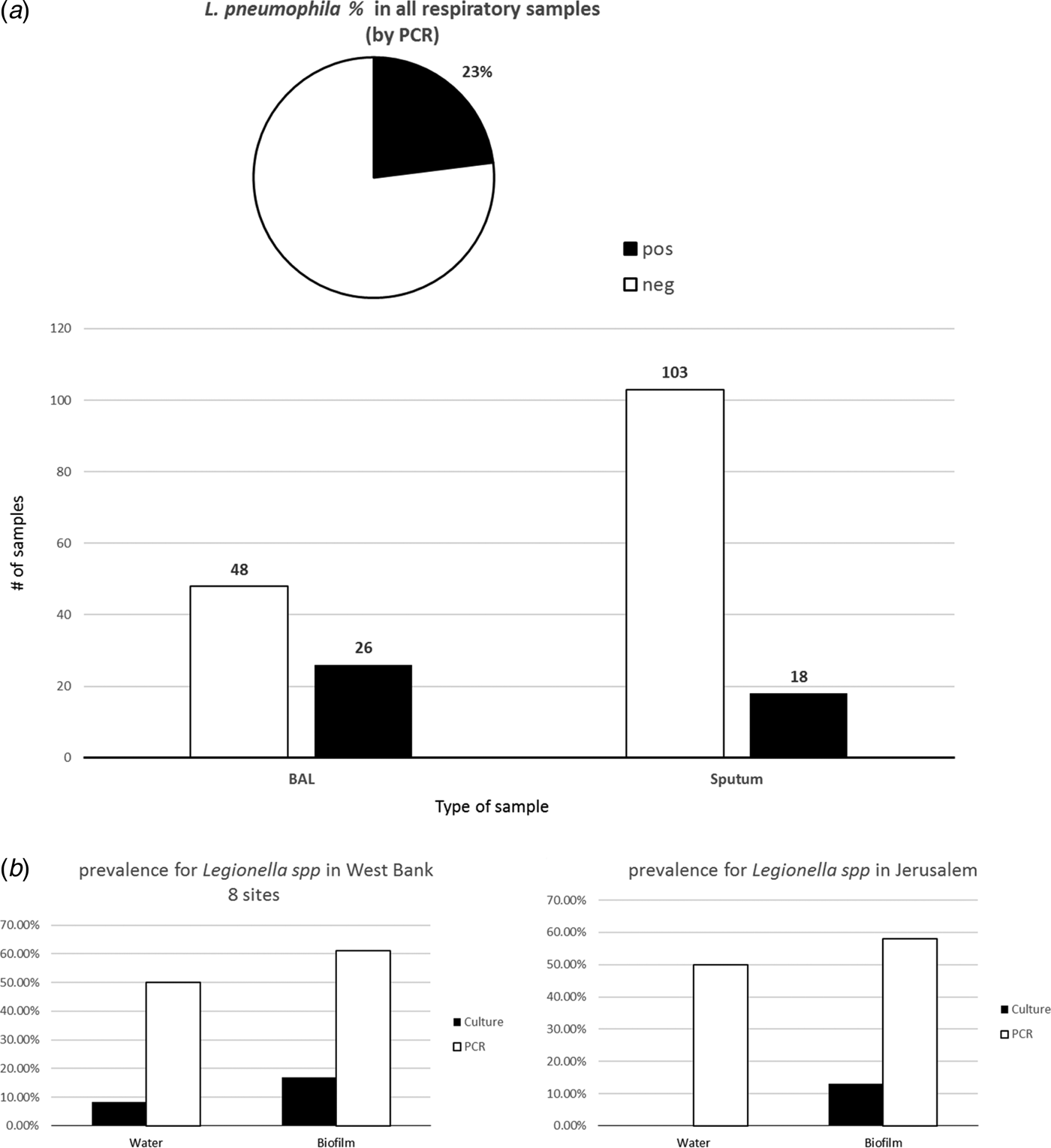
Fig. 2. (a) Prevalence of Legionella pneumophila in the respiratory samples (n = 121) collected from a hospital in Jerusalem as determined by PCR in DNA from BAL and sputum samples. (b) Prevalence of L. pneumophila from environmental samples of West Bank (eight sites) and a hospital in Jerusalem.
Comparison of allele profiles from respiratory and environmental samples
Out of 44 samples positive for L. pneumophila by PCR, 34 respiratory samples were selected and processed for NPSBT method according to the EWGLI standard scheme as explained in materials and methods. Gel electrophoresis of amplicons for the nested PCR of the 34 selected respiratory samples showed that 24 positive samples displayed at least four out of seven-allele products. The relevant bands of these 24 samples were sequenced. In addition, 15 environmental positives for L. pneumphila samples obtained from the different wards of the hospital were typed by NPSBT. The aim is to compare the STs obtained for the respiratory samples, with the STs of the environmental samples to find any nosocomial link. Of these environmental samples, 14 samples (positive for six- or seven-allele products) were sent to sequencing; one sample was only positive for one allele and excluded from further analysis.
The sequencing files were submitted to the respective website for quality check and identification of the corresponding STs. The following STs for respiratory samples were identified: ST 1 (29%, 7/24), ST 461 (21%, 5/24), ST 1037 (4%, 1/24) and 46% (11/24) undetermined profile (Fig. 3a). ST 1 was the most predominant ST in the respiratory samples. The results of STs of environmental samples revealed five different STs: ST 1 (28.5%, 4/14), ST 187 (21.4%, 3/14), one sample of ST 2070, ST 461 and ST 1482 (7.1%, 1/14), while the rest of the samples (28.5%, 4/14) was of unspecified STs (Fig. 3b). The most prevalent ST in the environmental samples was, like in the clinical samples, ST 1.
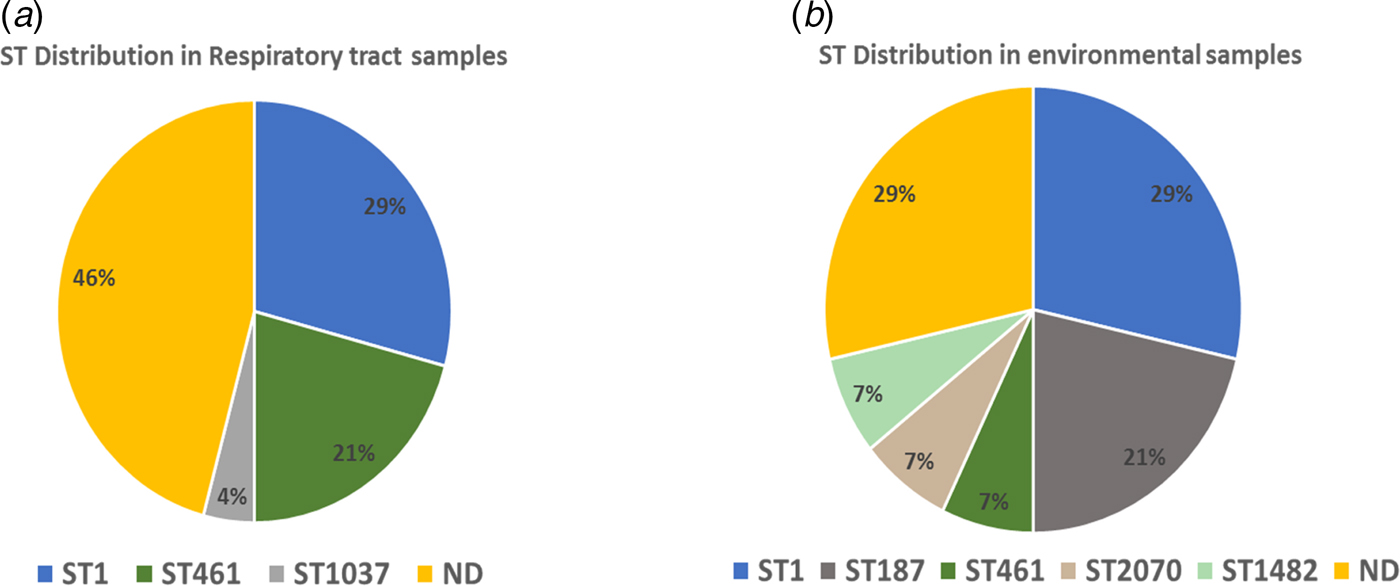
Fig. 3. Sequence types (STs) in clinical and environmental samples. ND = no ST identified. (a) ST distribution in respiratory tract samples. (b) ST distribution in environmental samples.
Discussion
Worldwide there has been a rising awareness regarding LD since it was first detected after an outbreak in 1976 in Philadelphia, USA. According to the recent European Center of Disease Prevention and Control (ECDC) report, 7022 cases were reported by 29 countries in the European Union during 2015 of which approximately 8% were fatal [Reference Beaute3]. However, in the West Bank, Palestine, LD is not a notifiable disease, and prior to this study, there was no previous record about the prevalence of Legionella spp. in environmental or respiratory samples. In a parallel study, we addressed the prevalence of Legionella spp. in drinking-water supply systems (DWSS) in eight major hospitals in the West Bank for a period of 3 years (2012–2015) and observed that all DWSS were contaminated with Legionella spp. (manuscript in preparation). Cultivation as well as molecular methods were applied in the current study and revealed a prevalence for Legionella spp. of 8.3% for water samples by culture, this percentage increased to 50% using PCR-based detection. As for biofilms, the Legionella prevalence was more evident, being 16.8% by culture vs. 61% by PCR analysis (data not shown).
Sputum samples collected from suspected pneumonia patients that were hospitalised at a hospital in East Jerusalem, in the period between September 2014 and June 2016. The age of the patients ranged between 3 months and 86 years. Patients were hospitalised in the following departments: internal medicine, ICU, paediatric, surgery paediatric ICU and gynaecology (Fig. 1). Paediatric patients showed a relatively high percentage of positive results which stresses further investigation or use of more advanced typing methods, like whole-genome sequencing (WGS) to decide whether the source of infection was nosocomial, or community-acquired. More surveillance steps should be taken.
Respiratory tract samples, including sputum and BAL samples, (n = 195) were collected from suspected pneumonia patients admitted to a hospital in East Jerusalem in the period between September 2014 and September 2015. This hospital is the only centre that can perform bronchoscopy to obtain BAL in the West Bank. Patients from all over the West Bank suspected of pulmonary infections needing bronchoscopy had their BAL samples extracted and examined in this medical centre. Therefore, we considered this centre to be representative for the whole of the population of the West Bank. The clinical samples (sputum n = 121, BAL n = 74) were cultured on agar medium selective for Legionella before DNA extraction. Only one sample was positive by the culture method [Reference Kirschner23, Reference Kozak24]. This very low yield of Legionella isolates from respiratory samples may be attributed to antibiotics being used prior to hospital admission.
In complementation to cultivation, we adopted molecular diagnostic approaches for Legionella infections which are implemented worldwide due to their high sensitivity, specificity and time efficiency [Reference Mercante and Winchell2]. Conventional PCR targeting the 16S rRNA gene was applied using DNA extracted directly from sputum and BAL samples collected from suspected pneumonia patients. Our results showed that 44 out of 195 samples (23%) of respiratory tract samples were positive for L. pneumophila, by conventional PCR. This indicates an annual incidence rate of 0.76 in 1 00 000 individuals, which coincides with many European countries, where LD has an overall incidence of 1.16 per 1 00 000 individuals of the EU. In Israel, a crude incidence of 0.67 cases/1 00 000 was reported in 2013 [Reference Moran-Gilad25].
In our study, BAL samples appeared to be sensitive and showed a higher percentage of positive PCR results (35%), whereas sputum samples showed lower per cent of positivity (15%) (Fig. 2a). Environmental samples (water and biofilms) in eight different hospitals in the West Bank besides Jerusalem were also abundant with Legionella spp. PCR detected a higher per cent of positive samples than culture-based detection (Fig. 2b). In addition to Legionella detection in clinical material, typing of Legionella strains is of considerable importance in determining sources of nosocomial legionellosis [Reference Qasem, Mustafa and Khan26]; therefore, we adopted NPSBT as a highly discriminatory typing method to provide useful information about the sources of infection particularly because it could be applied in situ where no Legionella isolates could be obtained.
In our study, we applied NPSBT to a set of clinical and presumptively linked environmental samples, i.e. the different hospital wards where the PCR-confirmed L. pneumophila patients were admitted in quest of any nosocomial link. Thirty-four PCR-positive respiratory specimens were tested by NPSBT using the protocol described previously [Reference Mentasti27]. After submission of the data files to the respective website, complete profiles (seven-allele) from the DNA of respiratory tract samples were obtained for 8.8% of the specimens, a further 53% resulted in five- or six-allele profile usually sufficient to identify the strain as belonging to one or two STs. A total of 17.6% of the samples gave three- or four-allele profile, which was sufficient to differentiate one profile from another without determining their ST precisely. Finally, 20.5% of the samples gave one- or two-allele profile for which we could not determine an ST. The STs and their distribution were as follows: ST 1 (32%), ST 461 (23%), ST 1037 (4%) and 46% undetermined allele profiles (Fig. 3a). ST 1 was the most predominant ST in the clinical samples. The results of the NPSBT analysis for the environmental samples (biofilm) revealed five different STs and their abundances as follows: ST 1 (28.5%), ST 187 (21.4%), one sample of each of ST 2070, ST 461 and ST 1482 (7.1%), while four samples contained unspecific STs. The most predominant ST in the selected environmental samples was also ST 1; this result is in agreement with our previous study which included eight hospitals in the West Bank; using MLVA for the genotyping of L. pneumophila isolates, the most prevalent genotype for our hospital as well as the other hospitals included in the survey was MLVA-genotype 4 which corresponds to ST 1 (Zayed et al., manuscript in preparation). On a global basis, ST 1 is the most predominant ST of L. pneumophila, [Reference Kozak-Muiznieks28] being more virulent than other STs [Reference Pancer29].
This study demonstrates the advantage of PCR-based approaches for sensitive detection and high-resolution identification of L. pneumophila in clinical samples, particularly, in a country where most patients use antibiotics prior to hospital admission; unfortunately, there is no record of the antibiotic usage prior admission to the hospital, as these patients are able to get an antibiotic without a medical prescription. Upon admission, after drawl of the BAL or sputum, patients would be under broad-spectrum antibiotics (mainly quinolones) as a prophylactic procedure before the bacteriology laboratory report. Such a situation makes it difficult to isolate L. pneumophila strains from clinical specimens.
Another important finding is the nosocomial health risk to susceptible patients, difficulty in relating clinical and environmental samples always stated [Reference Coscolla and Gonzalez-Candelas30–Reference Burillo, Pedro-Botet and Bouza32], since ST 1 was the most predominant ST in both clinical and environmental samples, and even though we cannot prove a direct link between the ST types from the clinical samples and the ST of the environmental samples. On the other hand, the occurrence of same L. pneumophila STs in the environmental samples poses a risk, and sustainable monitoring of hospital water systems is recommended [Reference Rodriguez-Martinez33]. Nowadays, WGS has been successfully applied for L. pneumophila providing the highest conceivable discriminatory power to distinguish outbreak from non-outbreak isolates [Reference Underwood34, Reference Bosch35]. To this end, further genomic and metagenomics studies are needed using DNA from the clinical and environmental samples used in this study [Reference Graham, Doyle and Jennison36].
Acknowledgements
This study was supported by a grant from the German Research Foundation (Deutsche Forschungsgemeinschaft DFG) grant number GZ: HO 930/5-1/2. The authors acknowledge Dr Amro Astal and the team in the hospital in Jerusalem for their cooperation in the collection of the clinical samples.



