


1. Introduction
Nowadays, Caenorhabditis elegans (C. elegans) is regarded as a model biological system, in which dozens of transcription factors (TFs) have now been identified and characterized by a combination of genetics, molecular screens and the genome sequencing project. Sufficient information for those TFs is available to suggest the important roles in cell determination, differentiation or other functions of them. However, only a small fraction of C. elegans TFs have so far been identified and studied on the candidate target genes and their expression patterns.
As we all know, the most basic step in the study of gene regulation is to uncover the target genes regulated by TFs. Recently, with the extensive application of chromatin immunoprecipitation followed by high-throughput sequencing technology (ChIP-seq), genome-wide identification of TF binding events becomes more facilitated. Nowadays, the numerous genome-wide TF binding data are freely available from the databases of ENCODE for human and mouse as well as modENCODE for C. elegans and Drosophila melanogaster, which may provide useful biological insights for understanding the mechanisms of transcriptional regulation. Nevertheless, the binding sites alone are not sufficient to infer regulation. Lots of previous studies were focused on identifying the target genes controlled by TFs and dissecting the transcriptional regulatory networks underlying the relevant biological process through a simple peak-based method (Boyer et al., Reference Boyer, Lee, Cole, Johnstone, Levine, Zucker, Guenther, Kumar, Murray, Jenner, Gifford, Melton, Jaenisch and Young2005; Chen et al., Reference Chen, Xu, Yuan, Fang, Huss, Vega, Wong, Orlov, Zhang, Jiang, Loh, Yeo, Yeo, Narang, Govindarajan, Leong, Shahab, Ruan, Bourque, Sung, Clarke, Wei and Ng2008; Kim et al., Reference Kim, Chu, Shen, Wang and Orkin2008). In those studies, the genes with promoters overlapping with one or more peaks were identified as the targets in most cases, but less information on the genes of other regions in genome was reported, especially for the regions out of 2 kb transcription start site (TSS) of a gene. In this case, some of the important long-range interactions were missed and it is unable to distinguish functional issues from non-functional binding sites. Furthermore, some of TF networks using ChIP-seq and gene expression data based on matrix decomposition have been constructed to unravel transcriptional regulatory programmes in complex systems (Yan et al., Reference Yan, Li, Yang, Shao, Jang, Guan, Zou, Van Waes, Chen and Zhan2013; Guan et al., Reference Guan, Shao, Deng, Wang, Zhao, Liang, Wang and Yan2014).
Meanwhile, gene expression profiles of more than 30 different cells and developmental stages as well as whole-animal RNA isolated from seven different developmental stages using tiling arrays have been generated to produce a spatial and temporal map of C. elegans gene expression (Spencer et al., Reference Spencer, Zeller, Watson, Henz, Watkins, McWhirter, Petersen, Sreedharan, Widmer, Jo, Reinke, Petrella, Strome, Von Stetina, Katz, Shaham, Ratsch and Miller2011). It provides a basis for establishing the roles of individual genes in cellular differentiation by measuring native transcripts from a broad array. The datasets of relative gene expression levels across tissue types and developmental stages are available online for further study.
It will be clear that the integration of the genome sequencing information and gene expression data in genome wide using C. elegans as a model biological system will result in uncovering the comprehensive transcriptional mechanisms. We hypothesized that a particular gene set regulated by a TF should behave in some measurably consistent manner. In this study, we tested the hypothesis by integrating TF binding and gene expression data of C. elegans in genome wide to further explore the mechanism of transcriptional regulation, which can be applied to other species.
2. Materials and methods
The summary of collected ChIP-seq and gene expression data was shown in Table 1. The workflow of gene expression and ChIP-seq data collection and integration analysis was shown in Fig. 1 and described as follows.

Fig. 1. The workflow of gene expression and ChIP-seq data collection and integration analysis.
Table 1. The summary of collected ChIP-seq and gene expression data
(i) ChIP-seq datasets collection
The ten ChIP-seq datasets of six TFs (HLH-1, ELT-3, PQM-1, SKN-1, CEH-14 and LIN-11) with tissue-specific and four TFs (CEH-30, LIN-13, LIN-15B and MEP-1) with broad expression patterns were collected from modENCODE database (Gerstein et al., Reference Gerstein, Lu, Van Nostrand, Cheng, Arshinoff, Liu, Yip, Robilotto, Rechtsteiner, Ikegami, Alves, Chateigner, Perry, Morris, Auerbach, Feng, Leng, Vielle, Niu, Rhrissorrakrai, Agarwal, Alexander, Barber, Brdlik, Brennan, Brouillet, Carr, Cheung, Clawson, Contrino, Dannenberg, Dernburg, Desai, Dick, Dose, Du, Egelhofer, Ercan, Euskirchen, Ewing, Feingold, Gassmann, Good, Green, Gullier, Gutwein, Guyer, Habegger, Han, Henikoff, Henz, Hinrichs, Holster, Hyman, Iniguez, Janette, Jensen, Kato, Kent, Kephart, Khivansara, Khurana, Kim, Kolasinska-Zwierz, Lai, Latorre, Leahey, Lewis, Lloyd, Lochovsky, Lowdon, Lubling, Lyne, MacCoss, Mackowiak, Mangone, McKay, Mecenas, Merrihew, Miller, Muroyama, Murray, Ooi, Pham, Phippen, Preston, Rajewsky, Ratsch, Rosenbaum, Rozowsky, Rutherford, Ruzanov, Sarov, Sasidharan, Sboner, Scheid, Segal, Shin, Shou, Slack, Slightam, Smith, Spencer, Stinson, Taing, Takasaki, Vafeados, Voronina, Wang, Washington, Whittle, Wu, Yan, Zeller, Zha, Zhong, Zhou, Ahringer, Strome, Gunsalus, Micklem, Liu, Reinke, Kim, Hillier, Henikoff, Piano, Snyder, Stein, Lieb and Waterston2010; Niu et al., Reference Niu, Lu, Zhong, Sarov, Murray, Brdlik, Janette, Chen, Alves, Preston, Slightham, Jiang, Hyman, Kim, Waterston, Gerstein, Snyder and Reinke2011; Landt et al., Reference Landt, Marinov, Kundaje, Kheradpour, Pauli, Batzoglou, Bernstein, Bickel, Brown, Cayting, Chen, DeSalvo, Epstein, Fisher-Aylor, Euskirchen, Gerstein, Gertz, Hartemink, Hoffman, Iyer, Jung, Karmakar, Kellis, Kharchenko, Li, Liu, Liu, Ma, Milosavljevic, Myers, Park, Pazin, Perry, Raha, Reddy, Rozowsky, Shoresh, Sidow, Slattery, Stamatoyannopoulos, Tolstorukov, White, Xi, Farnham, Lieb, Wold and Snyder2012). Briefly, the raw sequences were aligned to C. elegans genome (WS232) using ELAND allowing up to two mismatches per read. And peaks were called using PeakSeq with the threshold of P-value as 0·05 (Rozowsky et al., Reference Rozowsky, Euskirchen, Auerbach, Zhang, Gibson, Bjornson, Carriero, Snyder and Gerstein2009).
(ii) Target gene assignment and classification
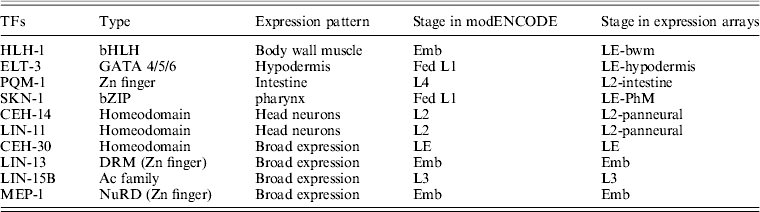
To identify candidate target genes possibly regulated by the TFs involved in the ChIP experiments, 5 kb regions both up- and downstream of a peak summit were first searched for transcriptional start (TSS) and end (TES) sites in the C. elegans genome (WS232) as described previously (Zhong et al., Reference Zhong, Niu, Lu, Sarov, Murray, Janette, Raha, Sheaffer, Lam, Preston, Slightham, Hillier, Brock, Agarwal, Auerbach, Hyman, Gerstein, Mango, Kim, Waterston, Reinke and Snyder2010). If no TSS was annotated for a gene, the TSSs were arbitrarily set to the positions 150 bases upstream of the translational start sites. A total of 31 640 protein-coding transcripts from 20 553 genes were used for target identifications. The targets of each ChIP-seq study were assigned to unique gene and the target genes were classified into 15 classes based on their positions relative to the peak summits. Those with peak summit within the gene region were defined as the class of C0; those with peak summit located 0–5 kb upstream the TSS were defined as C1–5 for each 1 kb interval, respectively, whereas 0–5 kb downstream the TES as C11–15, respectively (Fig. 2(a)). For those peaks with no candidate genes in the 5-kb flanking region, the two nearest genes were found. The nearest gene upstream the peak was defined as C21, and the second nearest was C22. Similarly, the nearest gene downstream the peak was defined as C23, and the second nearest was C24 (Fig. 2(b)). If the same gene could be assigned to several classes due to the different peaks nearby, it was assigned into a class with the smallest number (Fig. 2(c)). For example, if one gene could belong to both C1 and C4, it was defined as C1 target finally.
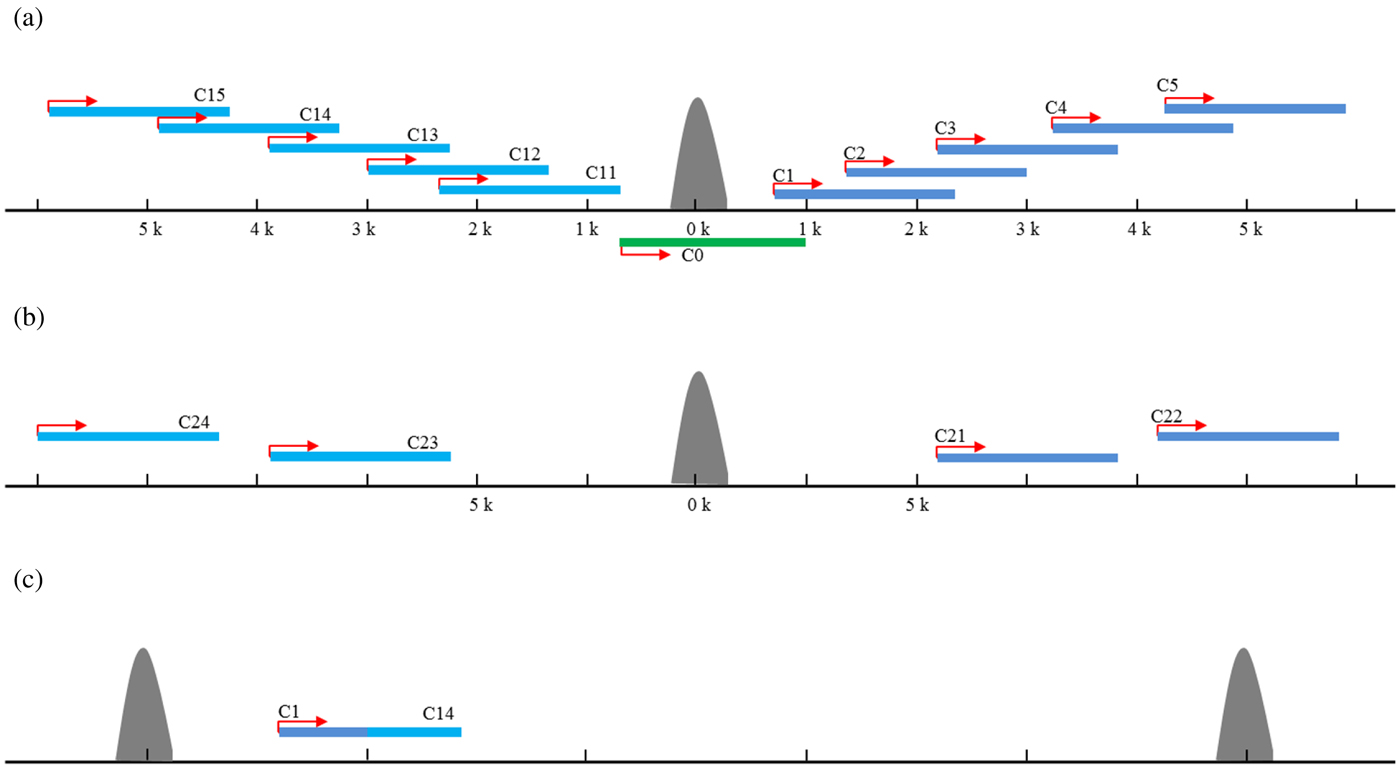
Fig. 2. The target genes assignment and classification. The targets of each ChIP-seq study were assigned to unique gene and the target genes were classified into 15 classes based on their positions relative to the peak summits. Those with peak summit within the gene region were defined as the class of C0; those with peak summit located 0–5 kb upstream the TSS were defined as C1–5 for each 1 kb interval, respectively, whereas 0–5 kb downstream the TES as C11–15, respectively (a). For those peaks with no candidate genes in the 5-kb flanking region, the two nearest genes were found. The nearest gene upstream the peak was defined as C21, and the second nearest was C22. Similarly, the nearest gene downstream the peak was defined as C23, and the second nearest was C24 (b). If the same gene could be assigned to several classes due to the different peaks nearby, it was assigned into a class with the smallest number (c).
(iii) Association of binding site locations with tissue-specific or broad expression
For association of the target genes of each TF with those enriched in tissue-specific cells or broad expression of whole animal based on microarray expression assays, expression values of all genes with corresponding patterns were derived from tiling array data (Spencer et al., Reference Spencer, Zeller, Watson, Henz, Watkins, McWhirter, Petersen, Sreedharan, Widmer, Jo, Reinke, Petrella, Strome, Von Stetina, Katz, Shaham, Ratsch and Miller2011). The summary of involved ChIP-seq and gene expression data were shown in Table 1. For each gene, the normalized log2 intensities from three replicates were averaged to give rise to its expression value. The expression values of the target genes were extracted for each gene. To plot the tissue-specific or broad expression of different classes of the target genes, those that all of whole genome genes were used as a reference and the plotting was performed with ‘vioplot’ package in R (Hintze JaN, Reference Hintze JaN1998). Mann–Whitney U-test was performed to calculate the significance levels (P-value) of gene expression difference between reference and the target genes of each class with cutoff of P-value as 0·001.
(iv) Calculation of TF activity
With both expression values and initial binding relationship provided, we can estimate transcription factor activity using proper mathematical model. Here, we applied network component analysis (NCA) method to calculate the transcription factor activity in 15 different classes for each TF (Liao et al., Reference Liao, Boscolo, Yang, Tran, Sabatti and Roychowdhury2003). Generally, the values for target genes were set to be 1 in initial transcription factor activity matrix, whereas all others were set to be 0. The final transcription factor activity was estimated by matrix decomposition, and the value will vary from 0 to some number bigger than 1.
3. Results and discussion
(i) Most of binding sites are located within 4 kb upstream of TSS
Based on the assignment and classification of ChIP-seq targets for all ten TFs, the target genes were classified into 15 classes, from C0 to C24 (Fig. 2, see in the Materials and methods). According to the proportion of target genes in each class of ten ChIP-seq studies, more than 60% of targets were classified from C0 to C4 (Fig. 3). It suggested that most of binding sites are located within 4 kb upstream of TSS, not only for the regulation of TFs with tissue-specific expression pattern but also for the regulation of TFs with broad expression pattern.

Fig. 3. The proportion of target genes in each class of ten ChIP-seq studies. The bar charts show the proportion of target genes in each class of ten ChIP-seq studies, including the TFs of HLH-1, ELT-3, PQM-1, SKN-1, CEH-14 and LIN-11 with tissue-specific expression patterns as well as CEH-30, LIN-13, LIN-15B and MEP-1 with broad expression patterns. X-axis represents the different ChIP-seq studies; Y-axis represents the percentage (%) of target genes in each class (C0–C24).
(ii) Integration analysis of binding site locations with tissue-specific expression data
Actually, there are several major transcriptional regulators of tissue-specific differentiation in C. elegans, which are expressed in specific tissues where they are known to bind and regulate diverse well-studied tissue-specific genes. Among the 22 TFs involved in the study of ChIP-seq as part of the modENCODE Consortium, there are some of tissue-specific TFs, including HLH-1 for body wall muscle (BWM), ELT-3 for hypodermis (Hyps), PQM-1 for intestine, SKN-1 for pharynx as well as CEH-14 and LIN-11 both for neurons (Table 1) (Niu et al., Reference Niu, Lu, Zhong, Sarov, Murray, Brdlik, Janette, Chen, Alves, Preston, Slightham, Jiang, Hyman, Kim, Waterston, Gerstein, Snyder and Reinke2011). Through read mapping with ELAND and peak calling with PeakSeq as well as the peak gene annotation, 13 384 target genes for HLH-1, 7989 target genes for ELT-3, 8337target genes for PQM-1, 10 553 target genes for SKN-1, 7759 target genes for CEH-14 and 4360 target genes for LIN-11 were identified, respectively. According to the binding site locations, the target genes were classified into 15 different categories (C0–C24) for all the six TFs studies (Table 2, see in Section 2).
Table 2. The number of target genes in each class for ten TFs
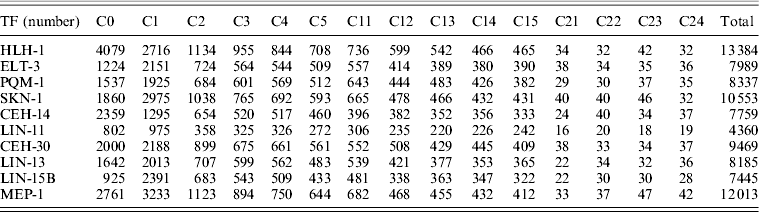
Numerous target genes have been identified by the ChIP-seq assays, but we are not certain whether the bindings are functional or not in vivo as well as whether the corresponding expression behaviours of each tissue-specific TF target genes are similar or not. We addressed the question by the integration analysis of the tissue-specific expression data derived from tiling arrays (Spencer et al., Reference Spencer, Zeller, Watson, Henz, Watkins, McWhirter, Petersen, Sreedharan, Widmer, Jo, Reinke, Petrella, Strome, Von Stetina, Katz, Shaham, Ratsch and Miller2011). The results of normalized expression levels in specific tissues for different categories of corresponding TF target genes, respectively, can be seen in Fig. 4. Moreover, the expression patterns of the mean expression distribution of each class in six tissue-specific studies were shown in Fig. 5(a). In common, all first three classes (C0–C2) of the target genes showed significantly elevated level of expression as opposed to that of reference whole genome genes for those six different TFs (Mann–Whitney U-test, P < 10−3), supporting that the majority of binding events are likely to be functional mostly through up-regulating its target genes within 2 kb upstream from TSS. Another class C3 was also reported as significantly elevated one in some of studies, including for ELT-3, SKN-1, CEH-14 and LIN-11. It indicated that bindings of those four TFs within 2–3 kb upstream from a gene's TSS may be also considered as one candidate group that more likely control its function by up-regulating its activities. Intriguingly, expression of all first six classes of the LIN-11 target genes representing genes with a peak inside (C0) or 0–5 kb upstream (C1–C5) from their TSS showed significantly higher than reference. In contrast, most classes of the 0–5 kb downstream (C11–C15) target genes for those six TFs showed decreased level of expression, suggesting a diverse expression behaviour of binding targets downstream from TSS that likely inhibit the TF activities.

Fig. 4. The expression patterns of tissue specific TFs target genes. The Violin plots of normalized expression levels in specific tissues for different categories of corresponding TF target genes were performed for six TFs with tissue-specific expression pattern, including HLH-1 for body wall muscle (BWM), ELT-3 for hypodermis (Hyps), PQM-1 for intestine, SKN-1 for pharynx as well as CEH-14 and LIN-11 both for neurons, respectively. X-axis represents 15 different classes of target genes and reference of genome gene expression; Y-axis represents the normalized gene expression value. Significant levels by Mann–Whitney U-test (P < 10−3) are indicated in the plots, dark-red for significantly up-regulated and dark-green for significantly down-regulated.

Fig. 5. The mean expression distribution and NCA results of each class in six tissue-specific studies. (a) It showed the mean expression distribution of each class in six tissue-specific studies, which is based on the integration analysis of ChIP-seq and gene expression data, including HLH-1 for body wall muscle (BWM), ELT-3 for hypodermis (Hyps), PQM-1 for intestine, SKN-1 for pharynx as well as CEH-14 and LIN-11 both for neurons, respectively. X-axis represents 15 different classes of target genes and reference of genome gene expression; Y-axis represents the mean level of normalized gene expression value. (b) It showed the transcription factor activity in 15 different classes for each of six TFs with tissue-specific expression pattern based on NCA method, including HLH-1 for BWM, ELT-3 for Hyps, PQM-1 for intestine, SKN-1 for pharynx as well as CEH-14 and LIN-11 both for neurons, respectively. X-axis represents 15 different classes of target genes and reference of genome gene expression; Y-axis represents the percentage of target genes with the corresponding A scores larger than 1 compared with the raw number of targets in ChIP-seq data.
Obviously, the mean expression level of target genes in C0 and C1 classes reached the summit as opposed to that of the other classes for all six TFs (Fig. 5(a)), which also suggested their most important roles in regulatory function. What is more interesting is that all of the class of C21 representing the nearest target genes out of 5 kb upstream from TSS showed sharply increased level of expression. However, the gene expression difference test compared with the reference is not significant, which is mainly due to the small sample size of genes count in C21.
In order to confirm our results, we have also performed NCA on the ChIP-seq and gene expression data. Compared to the traditional statistical methods of principal component analysis and independent component analysis, NCA is more comparable to determine low-dimensional representations of high-dimensional datasets for reconstruction of regulatory signals in biological systems (Liao et al., Reference Liao, Boscolo, Yang, Tran, Sabatti and Roychowdhury2003). Here, we applied NCA method to calculate the transcription factor activity in 15 different classes for each TF. According to the percentage of target genes with the corresponding A scores larger than 1 compared with the raw number of targets in ChIP-seq data, there appears to be maximum plot ratio in the class of C0, C1 and C21 (Fig. 5(b)). It suggests that they are three of most essential classes of target genes in the TF transcriptional regulation issues, which is consistent with our above integration analysis.
(iii) Integration analysis of binding site locations with broad expression data
Similarly, there are also several involved TFs with the broad expression pattern (expressed in the whole animal) in TF binding sites ChIP-seq database of modENCODE project, including CEH-30, LIN-13, LIN-15B and MEP-1. There were 9469 target genes for CEH-30, 8185 target genes for LIN-13, 7445 target genes for LIN-15B and 12 013 target genes for MEP-1, respectively, identified in those four broad expression TFs ChIP-seq. We then integrated all of the ChIP-seq targets information with the gene expression data in different whole animal developmental stages derived from the same tiling arrays (Spencer et al., Reference Spencer, Zeller, Watson, Henz, Watkins, McWhirter, Petersen, Sreedharan, Widmer, Jo, Reinke, Petrella, Strome, Von Stetina, Katz, Shaham, Ratsch and Miller2011). As expected, the bindings within 2 kb upstream of or within its target gene (C0–C2) showed significantly elevated level of expression compared to that of the reference for those four broad expression TFs (Mann–Whitney U-test, P < 10−3) (Fig. 6), which is the same as above results of six TFs with tissue-specific expression pattern. Moreover, similar results were observed for the bindings within 2–3 kb upstream (C3) of those TFs target genes except for MEP-1 as well as for the bindings within 3–4 kb upstream (C4) of LIN-15B. Generally, the bindings within 0–5 kb downstream (C11–C15) of those TFs showed constant or decreased expression values as opposed to reference, the significance appeared in C15 for LIN-13 and C5 to C15 for MEP-1. According to the mean expression distribution of each class in those four TF studies, the mean expression level of nearest bindings out of 5 kb upstream of target genes (C21) was also found to have a summit like C0 and C1 (Fig. 7(a)), which suggested its equally essential roles in regulatory function of TFs with broad expression pattern. The results were validated by the following NCA approach for those four TFs (Fig. 7(b)).

Fig. 6. The expression patterns of broad expressed TFs target genes. The Violin plots of normalized expression levels in whole animal for different categories of corresponding TF target genes were performed for four TFs with broad expression pattern, including CEH-30, LIN-13, LIN-15B and MEP-1, respectively. X-axis represents 15 different classes of target genes and reference of genome gene expression; Y-axis represents the normalized gene expression value. Significant levels by Mann–Whitney U-test (P < 10−3) are indicated in the plots, dark-red for significantly up-regulated and dark-green for significantly down-regulated.

Fig. 7. The mean expression distribution and NCA results of each class in four broad expression TFs studies. (a) It showed the mean expression distribution of each class in four broad expression TFs studies, which is based on the integration analysis of ChIP-seq and gene expression data, including CEH-30, LIN-13, LIN-15B and MEP-1, respectively. X-axis represents 15 different classes of target genes and reference of genome gene expression; Y-axis represents the mean level of normalized gene expression value. (b) It showed the transcription factor activity in 15 different classes for each of four TFs with broad expression pattern based on NCA method, including CEH-30, LIN-13, LIN-15B and MEP-1, respectively. X-axis represents 15 different classes of target genes and reference of genome gene expression; Y-axis represents the percentage of target genes with the corresponding A scores larger than 1 compared with the raw number of targets in ChIP-seq data.
(iv) Similar transcriptional regulation mechanisms of tissue-specific or broad expression transcription factors in C. elegans
Although diverse transcription factor binding features revealed by genome-wide ChIP-seq in C. elegans (Niu et al., Reference Niu, Lu, Zhong, Sarov, Murray, Brdlik, Janette, Chen, Alves, Preston, Slightham, Jiang, Hyman, Kim, Waterston, Gerstein, Snyder and Reinke2011), some of similar issues in regulatory mechanisms may be found between tissue-specific and broad expression TFs. Firstly, the majority of binding events in C. elegans are likely to be functional mostly through up-regulating its target genes, particularly three regions of within 2 kb upstream or within its target genes rather than those located elsewhere from its targets. Most of transcriptional regulation issues were previously reported to occur by a TF binding in a promoter region, which was defined as 2 kb from a TSS, particularly in simple organisms such as bacteria or yeast (Capaldi et al., Reference Capaldi, Kaplan, Liu, Habib, Regev, Friedman and O'Shea2008). However, the regulation within the target needs further validation. Secondly, some of regulation issues in C. elegans were also found to occur through long-range regions like enhancers, including 2–3 kb (C3) or 3–4 kb (C4) upstream from TSS. It looks similar as more complex species, in which more regulation occurs through long-range enhancers often spanning many tens of thousands of base pairs and the enhancer interactions become the principal form of gene regulation (Arnosti & Kulkarni, Reference Arnosti and Kulkarni2005; Lee et al., Reference Lee, Spilianakis and Flavell2005).
As we all know, transcription regulatory networks play a pivotal role in the development, function and pathology of metazoan organisms, which comprises direct or indirect protein–protein or protein–DNA interactions between TFs and their target genes. For our ten TFs, there existed synergistic effects or antagonistic effects between the TFs by co-binding with the same DNA, which have been reported in the previous studies (Lum et al., Reference Lum, Kuwabara, Zarkower and Spence2000; Vermeirssen et al., Reference Vermeirssen, Barrasa, Hidalgo, Babon, Sequerra, Doucette-Stamm, Barabasi and Walhout2007; Cheng et al., Reference Cheng, Yan, Hwang, Qian, Bhardwaj, Rozowsky, Lu, Niu, Alves, Kato, Snyder and Gerstein2011).
This work was funded by the Collaborative Research Fund (CRF) of Research Grants Council (RGC) in Hong Kong and Faculty Research Grants (FRG) from Hong Kong Baptist University as well as the Scientific Research Foundation and Academic & Technology Leaders Introduction Project, and 211 Project of Anhui University.
Declaration of interest
The authors declare that they have no competing interests.
Authors’ contributions
KH designed the study, collected the datasets from databases and analysed the data, then prepared the original draft the manuscript. DL, JS and ZZ helped to design the study and reviewed the manuscript. All authors read and approved the final manuscript.









