


1. Introduction
The aetiology of moderate to severe ID is highly heterogeneous. Mutations in over 450 genes have been implicated in ID, which only represents a minor part of all ID genes that are anticipated (Inlow & Restifo, Reference Inlow and Restifo2004; Schuurs-Hoeijmakers et al., Reference Schuurs-Hoeijmakers, Hehir-Kwa, Pfundt, van Bon, de Leeuw, Kleefstra, Willemsen, van Kessel, Brunner, Veltman, van Bokhoven, de Brouwer and de Vries2011; van Bokhoven, Reference van Bokhoven2011). The sheer number of ID genes makes identification of the primary genetic defect in individual families and isolated cases difficult. ID disorders show impressive phenotypic variability in their clinical presentation. A plethora of associated features can be seen in a range of ID syndromes. Other neurological features, such as autism, epilepsy, ADHD and behavioural anomalies are particularly common (van Bokhoven, Reference van Bokhoven2011), and for mutations in some genes, these features can be seen in various combinations or in isolation. Copy number variations (CNVs) predispose to a wide variety of syndromes associated with ID, neurodevelopmental disorders and psychiatric disturbances (Vissers et al., Reference Vissers, de Vries, Osoegawa, Janssen, Feuth., Choy., Straatman, van der Vliet, Huys, van Rijk, Smeets, van Ravenswaaij-Arts, Knoers, van der Burgt, de Jong, Brunner, van Kessel, Schoenmakers and Veltman2003; Sebat et al., Reference Sebat, Lakshmi, Malhotra, Troge, Lese-Martin, Walsh, Yamrom, Yoon, Krasnitz, Kendall, Leotta, Pai, Zhang, Lee, Hicks, Spence, Lee, Puura, Lehtimäki, Ledbetter, Gregersen, Bregman, Sutcliffe, Jobanputra, Chung, Warburton, King, Skuse, Geschwind, Gilliam, Ye and Wigler2007). Thus, microdeletions at 15q11·2, 15q13·3 and 16p13·11 can induce ideopathic generalized epilepsy (Helbig et al., Reference Helbig, Scheffer, Mulley and Berkovic2008), ID (Burnside et al., Reference Burnside, Pasion, Mikhail, Carroll, Robin, Youngs, Gadi, Keitges, Jaswaney, Papenhausen, Potluri, Risheg, Rush, Smith, Schwartz, Tepperberg and Butler2011), schizophrenia (Stefansson et al., Reference Stefansson, Rujescu, Cichon, Pietiläinen, Ingason, Steinberg, Fossdal, Sigurdsson, Sigmundsson, Buizer-Voskamp, Hansen, Jakobsen, Muglia, Francks, Matthews, Gylfason, Halldorsson, Gudbjartsson, Thorgeirsson, Sigurdsson, Jonasdottir, Jonasdottir, Bjornsson, Mattiasdottir, Blondal, Haraldsson, Magnusdottir, Giegling, Möller, Hartmann, Shianna, Ge, Need, Crombie, Fraser, Walker, Lonnqvist, Suvisaari, Tuulio-Henriksson, Paunio, Toulopoulou, Bramon, Di Forti, Murray, Ruggeri, Vassos, Tosato, Walshe, Li, Vasilescu, Mühleisen, Wang, Ullum, Djurovic, Melle, Olesen, Kiemeney, Franke, Sabatti, Freimer, Gulcher, Thorsteinsdottir, Kong, Andreassen, Ophoff, Georgi, Rietschel, Werge, Petursson, Goldstein, Nöthen, Peltonen, Collier, St Clair and Stefansson2008) and autism spectrum disorder (Pagnamenta et al., Reference Pagnamenta, Wing, Sadighi Akha, Knight, Bolte, Schmotzer, Duketis, Poustka, Klauck, Poustka, Ragoussis, Bailey and Monaco2009), but such microdeletions are also found in healthy individuals. There is a possibility that in different neurological disorders, the CNVs disturb the normal neurodevelopmental continuum by functioning to disrupt the homeostasis of neuronal development (Coe et al., Reference Coe, Girirajan and Eichler2012).
The NRXN1 gene is one of the largest human genes (1·1 Mb) with 24 exons, located on chromosome 2p16·3 (Boucard et al., Reference Boucard, Chubykin, Comoletti, Taylor and Sudhof2005). NRXN1 was previously recognized as a synapse protein in vertebrates and studies in Drosophila established that NRXN1 is present at the synapse where it regulates the levels of active zone protein bruchpilot (Zeng et al., Reference Zeng, Sun, Liu, Chen, Wei and Xie2007; Zweier et al., Reference Zweier, de Jong, Zweier, Orrico, Ousager, Collins, Bijlsma, Oortveld, Ekici, Reis, Schenck and Rauch2009). Neurexins are presynaptic cell-surface proteins that link the pre- and postsynaptic compartments of synapses by binding extracellularly to postsynaptic cell adhesion molecules and intracellularly to presynaptic PDZ domains (Missler et al., Reference Missler, Zhang, Rohlmann, Kattenstroth, Hammer, Gottmann and Südhof2003; Etherton et al., Reference Etherton, Blaiss, Powell and Sudhof2009). Nrxn1-knockout mice have defects in excitatory synaptic strength displayed in electrophysiological tests and show behavioural changes (Geppert et al., Reference Geppert, Khvotchev, Krasnoperov, Goda, Missler, Hammer, Ichtchenko, Petrenko and Südhof1998). Both point mutations and heterozygous microdeletions disrupting NRXN1 have been associated with a broad spectrum of psychiatric and neuronal development disorders including schizophrenia and autism. Compound heterozygous mutations in NRXN1 have been identified in syndromic ID in Pitt–Hopkins syndrome (Kirov et al., Reference Kirov, Gumus, Chen, Norton, Georgieva, Sari, O'Donovan, Erdogan, Owen, Ropers and Ullmann2008; Rujescu et al., Reference Rujescu, Ingason, Cichon, Pietilainen, Barnes, Toulopoulou, Picchioni, Vassos, Ettinger, Bramon, Murray, Ruggeri, Tosato, Bonetto, Steinberg, Sigurdsson, Sigmundsson, Petursson, Gylfason, Olason, Hardarsson, Jonsdottir, Gustafsson, Fossdal, Giegling, Möller, Hartmann, Hoffmann, Crombie, Fraser, Walker, Lonnqvist, Suvisaari, Tuulio-Henriksson, Djurovic, Melle, Andreassen, Hansen, Werge, Kiemeney, Franke, Veltman, Buizer-Voskamp, Sabatti, Ophoff, Rietschel, Nöthen, Stefansson, Peltonen, St Clair, Stefansson and Collier2009; Ching et al., Reference Ching, Shen, Tan, Jeste, Morrow, Chen, Mukaddes, Yoo, Hanson, Hundley, Austin, Becker, Berry, Driscoll, Engle, Friedman, Gusella, Hisama, Irons, Lafiosca, LeClair, Miller, Neessen, Picker, Rappaport, Rooney, Sarco, Stoler, Walsh, Wolff, Zhang, Nasir and Wu2010). In addition, compound heterozygous NRXN1 mutations consisting of a combination of exon-disrupting microdeletions and nonsense or splice-site mutations have recently been described in patients with severe early-onset epilepsy and profound ID (Harrison et al., Reference Harrison, Connell, Hayesmoore, McParland, Pike and Blair2011; Duong et al., Reference Duong, Klitten, Moller, Ingason, Jakobsen, Skjodt, Didriksen, Hjalgrim, Werge and Tommerup2012). Comorbid epilepsy has been reported in almost half of these cases (Ching et al., Reference Ching, Shen, Tan, Jeste, Morrow, Chen, Mukaddes, Yoo, Hanson, Hundley, Austin, Becker, Berry, Driscoll, Engle, Friedman, Gusella, Hisama, Irons, Lafiosca, LeClair, Miller, Neessen, Picker, Rappaport, Rooney, Sarco, Stoler, Walsh, Wolff, Zhang, Nasir and Wu2010; Gregor et al., Reference Gregor, Albrecht, Bader, Bijlsma, Ekici, Engels, Hackmann, Horn, Hoyer, Klapecki, Kohlhase, Maystadt, Nagl, Prott, Tinschert, Ullmann, Wohlleber, Woods, Reis, Rauch and Zweier2011).
In the current study we report a de novo microdeletion of ~455 kb affecting the NRXN1 gene in a patient with an ID phenotype and primary amenorrhoea due to gonadal dysgenesis. This is the first report of gonadal dysgenesis being associated with a NRXN1 deletion.
2. Materials and Methods
(i) Participants
Patient D1 was ascertained at the Department of Human Genetics of the Radboud University Medical Centre in Nijmegen, the Netherlands. She was included in a larger cohort study among individuals with unexplained ID.
(ii) Genotyping
Informed written consent was obtained prior to blood sampling from all members of affected families. Genomic DNA was extracted from the blood using a standard phenol–chloroform extraction method (Sambrook & Russell, Reference Sambrook and Russell2006) and the samples were stored at -20 °C until further processing. The affected and unaffected members of the D1 Dutch family were subjected to SNP array analysis using the Affymetrix 2·7 M cytogenetics array platform to obtain CNV data that revealed a heterozygous deletion of ~455 kb in patient D1. Subsequently the coding exons 2–24 of NRXN1 were sequenced by bidirectional sequencing using ABI-PRISM 3730 (Life Technologies, Foster City, CA, USA) as described previously (Gregor et al., Reference Gregor, Albrecht, Bader, Bijlsma, Ekici, Engels, Hackmann, Horn, Hoyer, Klapecki, Kohlhase, Maystadt, Nagl, Prott, Tinschert, Ullmann, Wohlleber, Woods, Reis, Rauch and Zweier2011). For the affected female D1, sequencing of the FSHR gene was also performed because of gonadal dysgenesis.
3. Results
(i) Patient report
Patient D1 (Fig. 1) is a female born to non-consanguineous Dutch parents. When referred for clinical genetic evaluation at age 28 years, mild ID was noted. The patient's mother had two abortions (both females) during early weeks of pregnancy. Patient D1 was born after a pregnancy of 42 weeks. The pregnancy was complicated by maternal hypertension, birth weight was low–normal (2980 g, 2·3th–5th percentile). Gross motor development was normal, but speech development was delayed, with the first single words being spoken after the age of 2 years. She went to a school for special education of children with developmental delay. Her behaviour was shy and introvert, and she was an anxious child. Medical problems included a primary amenorrhoea due to gonadal dysgenesis, diagnosed at the age of 16 years, which was treated with hormonal replacement therapy. Since the age of 16 years she has had recurrent episodes of loss of consciousness. Computed tomography of the brain and cardiologic evaluation did not reveal any abnormalities. Based on the clinical presentation of the events a probable psychogenic cause was considered. Upon physical examination she had a height of 173·5 cm (50th percentile), a weight of 70 kg (50th percentile) and a head circumference of 53·3 cm (39th percentile). She has minimal facial dysmorphic features including deeply set eyes and synophrys. Her feet were flat, long and narrow with long toes, and she also had long fingers. A metabolic screen could not explain the cause of her ID.
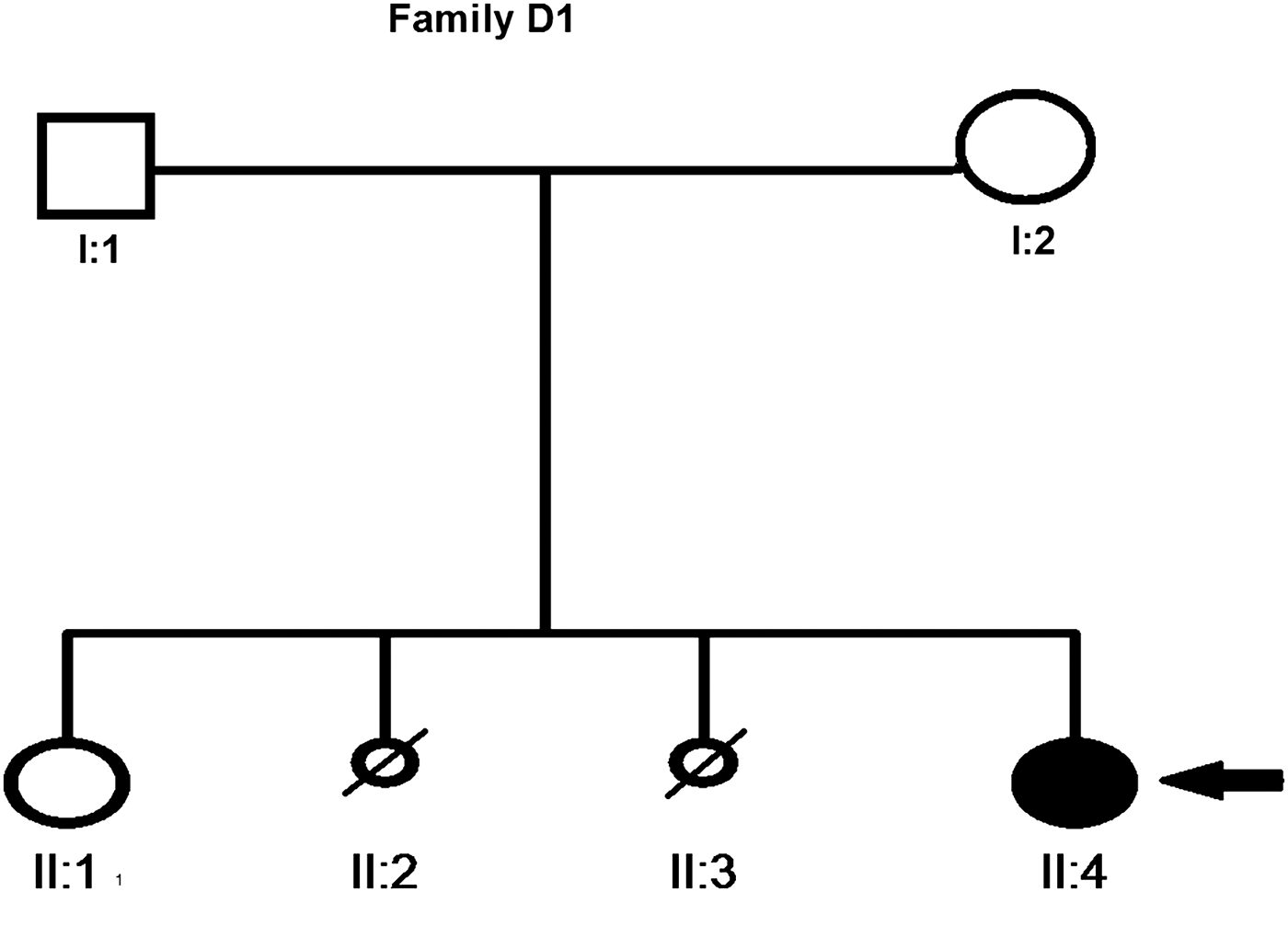
Fig. 1. Pedigree of family of patient D1. The small circles with diagonal lines represent the spontaneous abortion of female fetus while the filled circle shows the affected female.
SNP array analysis on the DNA of the patient with the Affymetrix 2·7 M cytogenetic array revealed an ~455 kb loss in the chromosomal region 2p16·3 (chr2: 51 020 477–51 476 031 Mb, Hg19, built February 2009), encompassing the alpha-promoter of NRXN1 including the first exon. Array analysis in both parents showed normal results, indicating that the deletion in this patient had occurred de novo (Fig. 2). Sequencing of the other allele of the NRXN1 gene of the patient did not reveal another pathogenic mutation in patient D1. The deletion abolishes exons 1–7 of the NRXN1 gene, similar to a large number of previously reported deletions. However, in contrast to the present patient, gonadal dysgenesis was not reported previously for any female deletion patients. Interestingly, the FSHR gene, which is associated with ovarian dysgenesis type I (OMIM 233300), is located proximal to the NRXN1 gene (Fig. 2). We hypothesized that the deletion might affect the expression of the FSHR gene, which together with a mutation on the other allele might be causative for the gonadal dysgenesis. However, sequence analysis of the FSHR gene in affected female D1 revealed only known polymorphisms but no mutation that could explain the gonadal dysgenesis.

Fig. 2. (a) Original picture of chromosomal region 2p16·3 carrying both NRXN1 and FSHR in close proximity. (b) Original data of CNV analysis derived from 2·7 Cytogenetic SNP array of patient D1 showing heterozygous deletion in the indicated chromosomal region 2p16·3 (chr2: 51 020 477–51 476 031 Mb, Hg19, built February 2009). (c) NRXN1 exons from 1–24, with its two isoforms (alpha-isoform and beta-isoform) as well as the ~455 kb microdeletion in patient D1, which overlaps exon 1–7 encoding for the alpha-isoform.
4. Discussion
We report a heterozygous de novo deletion of 455 kb that encompasses the upstream promoter and exons X–Y, encoding the alpha-isoform of NRXN1. A remarkable phenotypic variability has been observed for individuals with NRXN1 deletion, ranging from apparently unaffected carriers to individuals with severe cognitive deficits such as autism, epilepsy and schizophrenia. Recently, in a study conducted by Moller et al. (Reference Moller, Weber, Klitten, Trucks, Muhle, Kunz, Mefford, Franke, Kautza, Wolf, Dennig, Schreiber, Rückert, Wichmann, Ernst, Schurmann, Grabe, Tommerup, Stephani, Lerche, Hjalgrim, Helbig and Sander2013) on a cohort of individuals with idiopathic generalized epilepsy, a number of exon disrupting deletions as well as five index cases with heterozygous de novo changes in NRXN1 have been reported. Furthermore, two recent large studies by Dabell et al. (Reference Dabell, Rosenfeld, Bader, Escobar, El-Khechen, Vallee, Dinulos, Curry, Fisher, Tervo, Hannibal, Siefkas, Wyatt, Hughes, Smith, Ellingwood, Lacassie, Stroud, Farrell, Sanchez-Lara, Randolph, Niyazov, Stevens, Schoonveld, Skidmore, MacKay, Miles, Moodley, Huillet, Neill, Ellison, Ballif and Shaffer2013) and Bena et al. (Reference Bena, Bruno, Eriksson, van Ravenswaaij-Arts, Stark, Dijkhuizen, Gerkes, Gimelli, Ganesamoorthy, Thuresson, Labalme, Till, Bilan, Pasquier, Kitzis, Dubourgm, Rossi, Bottani, Gagnebin, Sanlaville, Gilbert-Dussardier, Guipponi, van Haeringen, Kriek, Ruivenkamp, Antonarakis, Anderlid, Slater and Schoumans2013) have reported an additional 34 and 25 pathogenic exon disrupting deletions in NRXN1, respectively. Recurrent clinical features associated with these deletions are ID, motor delay, autism, seizures, microcephaly and hypotonia. The patient from this study presents the main characteristic features that are observed in other deletion carriers. In addition, our patient shows gonadal dysgenesis. Amenorrhoea has not been reported previously in conjunction with NRXN1 deletions and it is possible that the gonadal dysgenesis might have another aetiology. However, it is noteworthy that the FSHR gene is the nearest neighboring gene of NRXN1 at the proximal side. Homozygous and compound heterozygous FSHR mutations are linked to ovarian dysgenesis (Aittomaki et al., Reference Aittomaki, Lucena, Pakarinen, Sistonen, Tapanainen, Gromoll, Kaskikari, Sankila, Lehväslaiho, Engel, Nieschlag, Huhtaniemi and de la Chapelle1995). Sanger sequencing of the FSHR gene in our patient did not reveal any DNA variants, which in conjunction with a positional effect of the deletion could give rise to the gonadal dysgenesis. Unfortunately, we were unable to test (bi-allelic) expression of FSHR in material from the patient and therefore it cannot be concluded whether close proximity of FSHR is coincidental nor exclude an involvement of the FSHR gene. The size and position of the NRXN1 deletion may determine whether or not FSHR expression is affected. There are only a few older females who have been investigated in the study of Bena et al. (Reference Bena, Bruno, Eriksson, van Ravenswaaij-Arts, Stark, Dijkhuizen, Gerkes, Gimelli, Ganesamoorthy, Thuresson, Labalme, Till, Bilan, Pasquier, Kitzis, Dubourgm, Rossi, Bottani, Gagnebin, Sanlaville, Gilbert-Dussardier, Guipponi, van Haeringen, Kriek, Ruivenkamp, Antonarakis, Anderlid, Slater and Schoumans2013). There was a remarkable excess of males (19 males vs. six females) and most females were prepubertal. Also the patients investigated in detail by Dabell et al. (Reference Dabell, Rosenfeld, Bader, Escobar, El-Khechen, Vallee, Dinulos, Curry, Fisher, Tervo, Hannibal, Siefkas, Wyatt, Hughes, Smith, Ellingwood, Lacassie, Stroud, Farrell, Sanchez-Lara, Randolph, Niyazov, Stevens, Schoonveld, Skidmore, MacKay, Miles, Moodley, Huillet, Neill, Ellison, Ballif and Shaffer2013) showed a male excess (20 males vs. 14 females) and all the females were prepubertal.
The study has a certain limitation, as sequencing of the other already identified genes correlated with ovarian dysgenesis was not performed due to the direct 2·7 K cytogenetic array approach but this technique cannot rule out the involvement of other probable genes causing ovarian dysgenesis in patient D1. Future case report studies should keep this limitation in their approach so that other probable causes can be sorted out before the use of higher techniques such as microarray and many others. In conclusion, the occurrence of gonadal dysgenesis presented here warrants longitudinal investigation of females with a NRXN1 deletion to establish a possible correlation with primary amenorrhoea.
We are grateful to all the family members for their participation in this study. This study was funded under the European Union's Seventh Framework Program under grant agreement number 241995, project GENCODYS. Z.A. was supported by the IRSIP program of the Higher Education Commission (HEC), Islamabad, Pakistan, and she also received funding from the COMSATS Institute of Information Technology for this work. C.Z. was funded by a grant from the Deutsche Forschungsgemeinschaft (Zw184/1-1). This study was also supported by funds from the Shifa College of Medicine and COMSATS Institute of Information Technology under a core grant to R.Q. as well as NRPU grant number 2155 from HEC.
Declaration of interest
None.


