





Crossref Citations
This article has been cited by the following publications. This list is generated based on data provided by Crossref.
Diep, Tram N.
Liu, Haoxin
and
Yan, Liang-Jun
2025.
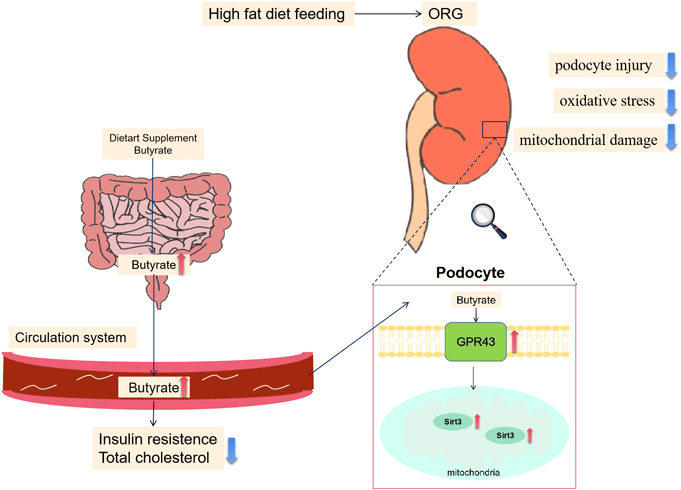
Beneficial Effects of Butyrate on Kidney Disease.
Nutrients,
Vol. 17,
Issue. 5,
p.
772.