


Introduction
A significant component of chemistry is the study of chemical reactions involving the cleavage of chemical bonds in reactants and the formation of chemical bonds in products. The reorganization of chemical bonds (or electron pairs) in a reaction originates from elevating the potential energy of the reactant molecules along their potential energy surface (PES) to form transient intermediates. When the energy of the transient intermediates approximates the conical intersection of the reactant’s PES and the product’s PES, the transient intermediates can relax along the product PES to dissociate old electron pairs and reassemble new electron pairs, yielding the products.
The initial electron energization in the reactants is the rate-limiting step of a chemical reaction. Heating the reactants thermalizes the chemical bonds (i.e., electron pairs), enabling the electrons to gain energy. The thermalization strategy usually energizes all electrons of the reactants at elevated temperatures, which may simultaneously activate multiple chemical bonds to form various products with undesirable selectivity. Delivering energy precisely to specific chemical bonds in reactants is the most promising way to drive chemical reactions with high product selectivity and energy utilization efficiency. Photochemistry, in which reactant molecules absorb light of a specific wavelength to activate chemical bonds for reaction, represents a successful example (Figure 1).

Figure 1. Schematic illustration of mechanisms responsible for weakening a chemical bond through photoexcitation of an electron that can fill the empty antibonding orbital of the chemical bond. The top process expresses the direct excitation of one electron from the bonding orbital to the empty antibonding orbital upon absorption of one photon. The bottom process describes an indirect strategy: electrons in a nanoparticle are first excited to the energy levels above its Fermi level (E F) upon light absorption, followed by the injection of one excited electron into the empty antibonding orbital of the chemical bond when the molecule is adsorbed on the surface of the nanoparticle. σ and σ* represent the bonding orbital filled with an electron pair and the empty antibonding orbital, respectively, of a sigma covalent chemical bond. In a complex molecule, the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) are usually used to describe the bonding orbital filled with electrons and the empty antibonding orbital, respectively.
The difference in quantum states between one photon and one free electron prevents the direct exchange of energy between individual photons and electrons. An ensemble of multiple electrons in atoms/molecules exhibits quantum states different from those of free electrons by forming unique band structures, within which electron–hole pairs can directly exchange energy with the appropriate light quanta. The excited electron–hole pairs dissociate into holes and energized electrons that can jump into the empty antibonding orbitals of molecules to weaken (or cleave) the corresponding chemical bonds (top, Figure 1). The limited number of electrons and energy states in single molecules limits the population of quantum states that can match the incident photons, restraining the light absorption power in the molecules. On the other hand, the population of quantum states matching photons of the incident light in a nanoparticle of a semiconductor or metal is significantly higher because of the increased number of electrons and energy states. As a result, nanoparticles usually exhibit a strong absorption of light over a broad spectral range, for example, visible light, which is of the greatest interest for research.
Light absorption in a nanoparticle excites electrons from the ground states to energy states higher than the Fermi level of the nanoparticle, resulting in the generation of so-called “hot electrons” and complementary “hot holes.” The hot electrons are no longer confined by individual atoms and valence bonds, thus becoming “free electrons,” which maximizes their capability of doing work using the energy gained from photoexcitation. When reactant molecules are adsorbed on the surface of a photoexcited nanoparticle, hot electrons can move across the nanoparticle surface and inject into the empty (antibonding) orbitals of the adsorbate molecules, facilitating energy exchange with the electrons in the adsorbate molecules to elevate the potential energy of the adsorbate molecules (bottom, Figure 1). The increased potential energy activates the adsorbate molecules to favor chemical reactions. In other words, filling hot electrons in the antibonding orbitals weakens the strength of the corresponding chemical bonds, favoring the cleavage of the bonds to promote chemical reactions. The role of hot electrons in mediating energy transfer from photons to reactant molecules endorses the terminology of “hot-electron chemistry.”Reference Linic, Aslam, Boerigter and Morabito1–Reference Buntin, Richter, Cavanagh and King4
The hot electrons lose energy to become cool ones after the activation of the reactant molecules. If the cool electrons flow back to the nanoparticle, they recombine with the hot holes to neutralize the charges, reverting the nanoparticle to its original state. If the cool electrons stay in the reactant molecules, the hot holes left in the nanoparticle are capable of driving oxidation reactions, supporting “hot-hole chemistry” and restoring the nanoparticle.Reference Thrall, Preska Steinberg, Wu and Brus5
In both scenarios, the nanoparticle likely remains intact, consistent with the characteristics of a catalyst. Thus, the chemical reactions triggered on photoexcited nanoparticles of semiconductors and metals could be described as “photocatalytic hot-carrier chemistry” to cover the reactions driven by both hot electrons and hot holes. Hot electrons exhibit mobility much higher than the complimentary hot holes, allowing the hot electrons (if generated) to inject into the target chemical bonds much faster than the hot holes traveling to the target adsorbate molecules. This difference determines the predominant role of “hot-electron chemistry” because “hot-hole chemistry” can only possibly happen after the consumption of the hot electrons in the “hot-electron chemistry.” As a consequence, extensive studies, including materials design and synthesis, in this emerging field focus on “hot-electron chemistry.”
Materials for hot-electron chemistry
Initiating hot-electron chemistry requires excitation of the ground-state electrons in catalysts to generate hot electrons, which must instantaneously inject into appropriate chemical bonds of the reactant molecules adsorbed on the catalyst surfaces, as highlighted in Figure 1 (bottom). The efficiency of generating hot electrons in a photoexcited catalyst nanoparticle mainly depends on the energy band structure and density of free electrons within the nanoparticle. On the other hand, the efficiency of injecting hot electrons into reactant molecules is primarily determined by the surface properties of the nanoparticle, including both the chemical activity and the physical properties (e.g., surface defects and geometrical curvature) that influence the spatial distribution of electrons and the adsorption of reactant molecules. The electron excitation dynamics in the former step and the hot-electron injection kinetics in the latter step must align and match to achieve the optimal photocatalytic efficiency, for example, the maximum turnover number of forming desirable products.
The rate constant of electron transfer from the nanoparticle surface to the surface adsorbate molecules across a distance, d, is expressed by:
 $${k_{{\rm{et}}}} = {1 \over h}{\left( {{{{{\rm{\pi }}^3}} \over {RT \cdot \Delta {E_{\rm{R}}}}}} \right)^{1/2}}H{_{{\rm{et}}}^{\rm{o}}^2}{e^{ - {\rm{\beta }}d}}{e^{ - {\Delta ^\ddag }G/RT}}$$,
$${k_{{\rm{et}}}} = {1 \over h}{\left( {{{{{\rm{\pi }}^3}} \over {RT \cdot \Delta {E_{\rm{R}}}}}} \right)^{1/2}}H{_{{\rm{et}}}^{\rm{o}}^2}{e^{ - {\rm{\beta }}d}}{e^{ - {\Delta ^\ddag }G/RT}}$$, in which 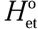 $H_{{\rm{et}}}^{\rm{o}}$ is the electronic coupling matrix element at d = 0, and ∆E R and ∆‡G are the reorganization energy and activation energy of adsorbate molecules, respectively, involved in the electron transfer process. The constants, h, R, and β are Planck’s constant, the universal gas constant, and a parameter measuring the sensitivity of the electronic coupling matrix element to distance, respectively. The adsorption strength of reactant molecules that determines d and the adsorption configuration that determines ∆E R are crucial to tune the electron transfer rate. For instance, adsorption of a specific reactant on widely used industry catalysts usually exhibits a high adsorption strength and an appropriate adsorption configuration, corresponding to a small d and a low ∆E R, both of which favor fast electron-transfer kinetics. Therefore, industry catalysts could be the starting point for developing efficient photocatalysts for hot-carrier chemistry with a focus on engineering the internal structures of the catalyst particles to realize the efficient generation of hot electrons and proper matching with their injection kinetics.
$H_{{\rm{et}}}^{\rm{o}}$ is the electronic coupling matrix element at d = 0, and ∆E R and ∆‡G are the reorganization energy and activation energy of adsorbate molecules, respectively, involved in the electron transfer process. The constants, h, R, and β are Planck’s constant, the universal gas constant, and a parameter measuring the sensitivity of the electronic coupling matrix element to distance, respectively. The adsorption strength of reactant molecules that determines d and the adsorption configuration that determines ∆E R are crucial to tune the electron transfer rate. For instance, adsorption of a specific reactant on widely used industry catalysts usually exhibits a high adsorption strength and an appropriate adsorption configuration, corresponding to a small d and a low ∆E R, both of which favor fast electron-transfer kinetics. Therefore, industry catalysts could be the starting point for developing efficient photocatalysts for hot-carrier chemistry with a focus on engineering the internal structures of the catalyst particles to realize the efficient generation of hot electrons and proper matching with their injection kinetics.
Figure 2 compares hot-electron generation in typical photocatalysts made of semiconductors and metals. Absorbing light of appropriate wavelengths in a semiconductor excites electrons from the valence band (VB) to the conduction band (CB) to generate hot electrons (Figure 2a). In contrast, the existence of high-density ground-state free electrons and the overlap of the VB and CB in a metal promote broadband light absorption to generate large populations of hot electrons (Figure 2b). The density of hot electrons in an excited semiconductor is usually much lower than in an excited metal due to the limitation of many parameters such as dopant concentration, covalent bond strength, bandgap, and light absorption coefficient. Low density of hot electrons limits the population of hot electrons with the quantum states matching the empty orbitals in the adsorbate reactant molecules, restraining hot-electron injection. However, the lifetime of hot electrons in a semiconductor is much longer than in a metal (i.e., nanosecond-picosecond for semiconductor versus femtosecond for metal), which offers more time for the adsorbate molecules to reorganize their adsorption configuration to match the quantum states of the hot electrons, promoting the occurrence of hot-electron injection. Despite the feasibility of exciting high-density hot electrons in a metal, they can loss their energy rapidly because of their short lifetime, discouraging hot-electron injection (detailed explanations are given in the Yu et al.Reference Yu, Mohan and Jain6 and Harutyunyan et al.Reference Harutyunyan, Suchanek, Lemasters and Foley IV7 articles in this issue). The distinct characteristics of hot electrons generated in semiconductors and metals represent the driving force to design and synthesize new materials, which promote the positives and suppress the negatives, for photocatalysis in the past decades.

Figure 2. Schematic illustration of hot-electron distribution in photoexcited nanoparticles of different material systems: (a) semiconductor, (b) metal, (c) semiconductor–metal composite with metal as the light absorber, and (d) metal–semiconductor composite with semiconductor as the light absorber. The arrows highlight the flow direction of hot electrons from the photocatalyst nanoparticles to the empty lowest unoccupied molecular orbital (LUMO) of molecules adsorbed on the surfaces of the nanoparticles. The blue color indicates cool electrons (i.e., electrons with energy below or near the Fermi level, E F, of the materials). The warming colors indicate hot electrons with energy above the Fermi level of the materials. Note: CB, conduction band; VB, valence band; HOMO, highest occupied molecular orbital.
The synthesis of composite nanostructures integrating plasmonic metal nanoparticles and wide-bandgap semiconductor nanoparticles represents a promising example of promoting the positives ad suppressing the negatives that has emerged in recent years since the plasmonic metal nanoparticles exhibit strong absorption of visible light to generate hot electrons. The short lifetimes of hot electrons in metal nanoparticles can be alleviated by transferring them to the CB of the interfaced semiconductor nanoparticles (Figure 2c). The Schottky barrier at the (n-type) semiconductor–metal interface prevents the backflow of electrons. The extended lifetime of electrons in the semiconductor’s CB allows the accumulation of hot electrons to a high population, benefiting the hot electron injection to the reactant molecules adsorbed on the semiconductor surface. In this issue, Huang et al. overview the promise and applications of this class of photocatalysts.Reference Huang, Guo, Hu and Wei8
Hot-electron flow from metal nanoparticles to a semiconductor can be probed with nanodiode reactors, a unique design pioneered by Somorjai and co-workers.Reference Ji, Zuppero, Gidwani and Somorjai9 The working principle and applications are discussed in detail by Park and Somorjai in this issue.Reference Park and Somorjai10 Similar to the pure semiconductor photocatalysts shown in Figure 2a, the major challenge of a metal/semiconductor catalyst shown in Figure 2c is the limited types of reactant molecules that can be strongly adsorbed on the semiconductor, restraining the scope of photocatalytic reactions. This challenge can explain why metal–semiconductor composite catalysts have been mostly synthesized by following the design principle shown in Figure 2d.Reference Wang, Li and Xu11 The semiconductor component absorbs light to generate hot electrons while the metal component receives hot electrons to drive interesting chemical reactions on the metal surface, on which a broad range of reactants can be adsorbed by tuning the metal composition. The Schottky barrier at the semiconductor–metal interface also blocks electron flow from the metal back to the semiconductor. The low density of hot electrons in the semiconductor and the short lifetime of hot electrons in the metal simultaneously prevent the accumulation of high-density hot electrons in the metal, which limits the maximum energy of the hot electrons in the metal. Nontraditional semiconductors have also been explored as light antennae to replace the traditional inorganic semiconductors for photocatalysis.Reference Wang, Vogel, Sachs, Sprick, Wilbraham, Moniz, Godin, Zwijnenburg, Durrant, Cooper and Tang12 Wang et al. present a story of self-assembled porphyrin solids in this issue.Reference Wang, Fan and Bai13
The use of metal–semiconductor composite photocatalysts can mitigate the challenges of either the metals or the semiconductors, but not both. This composite design not only complicates materials synthesis, but also initiates new challenges associated with the newly formed metal–semiconductor interfaces. Examples include possible low electron transfer efficiency across the interfaces, and unsatisfactory robustness of the interface. Therefore, it becomes more promising to engineer nanoparticle photocatalysts of either metals or semiconductors to alleviate their challenges while fully maintaining their meritorious properties. For instance, nanoparticles of numerous transition metals and metal alloys can strongly adsorb reactants of many important reactions to serve as widely used catalysts in both industry and research. However, the short lifetimes of hot electrons in photoexcited metal nanoparticles represents the fundamental barrier to prevent them from being used as photocatalysts for hot-electron chemistry. Reducing the travel distance of hot electrons in small metal nanoparticles and promoting the hot-electron injection kinetics from the metal nanoparticles to the adsorbate reactants can simultaneously lower the requirement of extended lifetime of hot electrons and thus benefit photocatalytic hot-electron chemistry using pure metal nanoparticles. Sun and coworkers have highlighted the potential of reducing the size of metal nanoparticles to a region (usually <10 nm), at which the surface quantum phenomenon of Coulomb blockade (i.e., electrons in small-enough objects creating strong Coulomb repulsion) occurs to favor hot-electron injection, to promote efficient hot-carrier chemistry on the so-called “quantum-sized metal nanoparticles.”Reference Wei, Wu and Sun14
Thermodynamic and kinetic considerations
Injecting hot electrons into the empty orbitals of reactant molecules adsorbed on the surface of a photocatalyst represents the prerequisite to drive “hot-electron chemistry,” in which the reaction thermodynamics and kinetics are different from those of the corresponding chemical reaction driven thermally. If the injected electrons stay in the adsorbate molecules until the molecules desorb from the photocatalyst, the reactant molecules become negatively charged radicals with tuned chemical reactivity (e.g., higher reactivity in general) to involve in the desired reaction. Such a change in reactivity will alternate both the thermodynamic and kinetic diagrams of the chemical reaction. For example, selective oxidation of primary alcohol to aldehyde with ambient-pressure oxygen molecules (O2) is kinetically difficult at room temperature, although it is thermodynamically spontaneous (or exergonic).Reference Hermes, Janes and Schmidt15 The slow oxidation kinetics can be overcome in the presence of silica-supported platinum (Pt) nanocrystals under photoillumination.Reference Zhang, Han, Xu, Foley IV, Zhang, Codrington, Gray and Sun16 Photoexcitation of the Pt nanocrystals generates hot electrons that are injected into O2 adsorbed on the Pt surface, leading to the formation of freestanding superoxide radicals (O2•−). The increased oxidizing power of O2•− enables fast oxidation of benzyl alcohol to benzaldehyde due to the lowered activation energy barrier. The leftover holes in the Pt nanocrystals also possess strong oxidizing power to drive the oxidation of benzyl alcohol on the Pt surface. The new reactions initiated by hot electrons and hot holes should be synergistically compatible to maximize the efficiency of hot-carrier chemistry.
Another common scenario is that hot electrons are not retained in adsorbate reactant molecules after their energy is deposited to the reactant molecules (Figure 3). A typical exergonic reaction with reaction Gibbs energy of ΔrG < 0 is thermodynamically spontaneous to convert reactant to a product when the ground-state reactant is thermalized to increase its energy by E a (or forward reaction activation energy) along its PES. Such an energy elevation transforms the reactant to a transient intermediate that can cross the conical intersection to fall on the product’s PES. Relaxing the intermediate along the product’s PES to the ground state forms the stable product with releasing energy (black curve, Figure 3a). The value of E a determines the forward reaction rate of converting the reactant to the product according to the Arrhenius equation. A smaller E a allows a faster reaction at a given temperature. When a reactant molecule adsorbed on the surface of a photoexcited metal nanoparticle receives a hot electron, the reactant molecule becomes a negatively charged transient intermediate (NCTI) with a PES of higher energy (red curve, Figure 3a).Reference Linic, Aslam, Boerigter and Morabito1 The injected hot electron exchanges energy with other electrons in the NCTI until the hot electron loses its energy to become a lukewarm one and the NCTI oscillates near its PES valley. The lukewarm electron then flows back to the metal nanoparticle, and the NCTI relaxes back to the reactant at an energy level (R*, Figure 3a) higher than the ground state of the reactant by following the Franck–Condon principle, which is a rule in quantum chemistry explaining the intensity of vibronic transitions. These sequential steps lower the activation energy of the forward reaction from E a to E a* (Figure 3a), favoring the reaction kinetics to accelerate the forward reaction.

Figure 3. Energy diagrams (black curves) of (a) an exergonic reaction and (b) an endergonic reaction, showing the requirement of activation energy in the reactant (R) to overcome the forward reaction energy barrier (E a). Injection of hot electrons into the reactant molecules forms negatively charged transient intermediate (NCTI) species, which exhibit potential energy surface (PES) (red curves) significantly different from the reactant. Depending on the position of the PES valley of the NCTI, relaxing the NCTI to a ground state either (a) lowers the forward reaction energy barrier (E a*) or (b) directly forms the product.
If the PES valley of the NCTI is above the product’s PES, flowing the lukewarm electron back to the metal nanoparticle will relax the NCTI to form product directly (Figure 3b). This process represents a feasible paradigm to crack down on the thermodynamic nonspontaneity of an endergonic reaction with reaction Gibbs energy of ΔrG > 0, enabling the conversion of reactant to product. In this case, the light quanta can be considered as a type of massless reactant to react with the existing reactant. The photon energy elevates the total Gibbs energy of all the reactants to a level higher than the Gibbs energy of the product, realizing the thermodynamic flip as shown in Figure 3b. In both paradigms highlighted in Figure 3, hot electrons in the metal nanoparticle catalysts behave more like mediators to facilitate energy transfer from photons to the desirable chemical molecules, thus significantly changing both the kinetics and thermodynamics of the reactions.
Challenges and outlook
Controlling the flow dynamics of hot electrons is crucial to driving hot-electron chemistry on the surfaces of selected catalysts efficiently. In a photoexcited catalyst particle, the desirable directional flow of hot electrons competes with many other electron relaxation pathways, including electron–lattice collision, electron–surface collision, and electron–hole recombination. These competitive relaxation processes convert the kinetic energy of hot electrons to heat in the catalyst particle (the so-called photothermal effect), undoubtedly draining the power of hot electrons toward hot-electron chemistry. The heat elevates the temperature of the catalyst, reactant, and product to increase the reaction rate according to the Arrhenius equation, which seems equivalent to the increased reaction rate driven by the hot-electron chemistry as shown in Figure 3a. Such a similarity of the photothermal effect and hot-electron chemistry (or so-called nonthermal effect) in accelerating reaction kinetics makes it challenging to distinguish their contributions in the chemical reactions involving single reaction pathways.Reference Yu, Mohan and Jain6,Reference Zhou, Swearer, Zhang, Robatjazi, Zhao, Henderson, Dong, Christopher, Carter, Nordlander and Halas17–Reference Zhang, Li, Reish, Zhang, Su, Gutiérrez, Moreno, Yang, Everitt and Liu20
However, the photothermal effect and nonthermal effect can behave differently in determining product selectivity of the chemical reactions involving multiple reaction pathways. The photothermal effect accelerates the reaction rate of all pathways, making it difficult to improve the product selectivity significantly. By contrast, the directional flow of hot electrons to the target chemical bonds of reactant molecules mainly accelerates the reaction pathway of forming the product that requires the activation of the target bonds. The product formation rate of other reaction pathways is influenced much less. Therefore, nonthermal hot-electron chemistry offers a promising strategy to improve product selectivity for complex chemical reactions. Since the photothermal effect is inevitable, maintaining a constant temperature of a reaction system by using appropriate controls (e.g., water bath or fast gas flow) to remove heat from the reaction system swiftly becomes critical to maximize the improvement of product selectivity enabled by the hot-electron chemistry. Moreover, reducing the size of catalyst nanoparticles could lower the electron–lattice collision frequency (i.e., the dominant process responsible for heat generation) in individual nanoparticles to suppress the photothermal effect. The travel time of hot electrons in the small catalyst nanoparticles becomes short, allowing more hot electrons to reach the nanoparticles’ surface on which hot-electron injection occurs before they relax back to the ground state.
The great promise of nonthermal hot-electron chemistry in improving product selectivity relies on control over the flow direction of hot electrons to the target chemical bonds of reactant molecules adsorbed on the catalyst surface. In general, the antibonding orbitals with a high local density of states (LDOS) and low energy are preferable to receive hot electrons, leading to the selective activation of the corresponding chemical bonds in the reactant molecules. According to Equation 1, a short distance from the catalyst surface to the target chemical bonds is necessary to achieve rapid hot-electron injection. Therefore, appropriate adsorption configuration of the reactant molecules on the catalyst surface is essential to determine the flow dynamics and flow direction of hot electrons. Theoretical modeling and calculations such as density functional theory can play an important role to help comprehensively understand the adsorption configuration of reactant molecules on the catalyst surfaces and the energy-dependent LDOS. The calculation results provide guidance for designing the optimum catalysts with the appropriate surfaces to adsorb reactant molecules in the desirable configurations, which favor the injection of hot electrons into the preferable chemical bonds in the adsorbed reactant molecules.
The competitive relaxation processes of hot electrons undoubtedly drain the ability of hot electrons in driving hot-electron chemistry on the surface of the catalyst nanoparticles. The process with the fastest dynamics (corresponding to the shortest characteristic transient time) dominates energy dissipation of the hot electrons. As a result, achieving a high-energy-conversion efficiency of hot-electron chemistry prefers hot-electron injection with the shortest transient time along with elongated transient times for the other competitive processes. Measuring the dynamics of the transient processes involved in hot-electron relaxation necessitates comprehensive understanding of the behavior of hot electrons in the photoexcited catalyst nanoparticles as well as on the nanoparticles’ surfaces.Reference Harutyunyan, Suchanek, Lemasters and Foley IV7 Despite the challenges, it is essential to probe the transient processes of hot electrons using the currently available transient spectroscopy, including time-resolved emission spectroscopy, transient optical absorption spectroscopy, transient x-ray absorption spectroscopy, and transient Raman spectroscopy. Special attention is necessary to carefully adjust the intensity of the pump probes to avoid damage to the catalysts and direct photolysis of reactant/product molecules. Integrating these spectroscopic data will provide direct information to compare the dynamics of individual transient processes of hot electrons. The optimum catalyst should possess a rapid hot-electron injection kinetics with a characteristic time much shorter than the characteristic times of other competitive transient processes.
In summary, photocatalytic hot-electron chemistry is mechanistically different from conventional thermal chemistry, representing a promising strategy to accelerate reactions, improve product selectivity, and maximize energy conversion efficiency. Small-sized nanoparticles of transition metals that are widely used as catalysts in industry and research could be directly photoexcited to drive efficient hot-electron chemistry, which represents an immediate interest to explore.Reference Zhang, Li, Reish, Zhang, Su, Gutiérrez, Moreno, Yang, Everitt and Liu20–Reference Dai, Wei, Duong and Sun23 The light absorption power in the small nanoparticles is usually too low to efficiently harvest photon energy. Integrating the catalyst nanoparticles with light antennae that can concentrate the incident light to generate enhanced local electric fields represents a promising solution to maximize the light-harvesting efficiency and a promising research direction. For example, plasmonic nanostructuresReference Swearer, Zhao, Zhou, Zhang, Robatjazi, Martirez, Krauter, Yazdi, McClain, Ringe, Carter, Nordlander and Halas24 and geometrically symmetric dielectric spheresReference Dai, Rasamani, Hall, Makrypodi and Sun25 represent two typical classes of light antennae that rely on localized surface plasmon resonances and surface light-scattering resonances, respectively, to generate localized strong electric fields near their surfaces. Composites of small catalyst nanoparticles on nanostructured light antennae could improve their light absorption power to enhance their photocatalytic activity. A comprehensive understanding of the mechanism of photocatalytic hot-electron (hole) chemistry will benefit the decades-long, but recently renascent, research field by advancing the materials design and synthesis of photocatalysts.

Yugang Sun is an associate professor of chemistry at Temple University. He received his BS and PhD degrees from the University of Science and Technology of China in 1996 and 2001, respectively. From 2006 to 2015, he was a research scientist at the Center for Nanoscale Materials at Argonne National Laboratory. He received the Presidential Early Career Award for Scientists and Engineers in 2007 and the US Department of Energy Office of Science Early Career Scientist and Engineering Award in 2008. He is a highly cited materials scientist and chemist. His research focuses on the design/synthesis of functional nanomaterials, the development of in situ synchrotron x-ray techniques, and catalysis. Sun can be reached by email at [email protected].

Zhiyong Tang is a professor with the National Center for Nanoscience and Technology, China. He obtained his bachelor’s and master’s degrees from Wuhan University, China, in 1993 and 1996, respectively. He obtained his PhD degree in 1999 at the Changchun Institute of Applied Chemistry, Chinese Academy of Sciences. He completed postdoctoral research at the Swiss Federal Institute of Technology Zürich, Oklahoma State University, and the University of Michigan. He was awarded the 100 Talents Program, Chinese Academy of Sciences, in 2006. Tang can be reached by email at [email protected].



