



Introduction
The mineral magnetoplumbite was described by Aminoff (Reference Aminoff1925) from the Långban iron-manganese mines, Värmland County, Sweden. The formula and the topology of the crystal structure was first correctly interpreted by Adelsköld (Reference Adelsköld1938). The composition of this archetypal mineral is given as ideally Pb[Fe12]O19. It is isostructural with Ba[Fe12]O19 (barioferrite), a common synthetic permanent magnetic material (e.g. Pullar, Reference Pullar2012). They both belong to a wider family of compounds, the so-called hexagonal ferrites (or hexaferrites). The group members (Table 1) are rare as minerals, but are found in a variety of geological environments, including metasomatic skarns, high-grade metamorphic rocks (granulites), kimberlites, lherzolites, lamproites, volcanic and pyrometamorphic rocks and chondritic meteorites, altogether indicating significantly wide P–T–f O2 stability conditions for the structure type. The minerals of the group, all possessing basic hexagonal crystal symmetry, are described by the general formula AB 12O19, where A is a large cation (A 2+ or A 1+) and B usually represents more highly charged cations of intermediate size. In the present paper, we announce the newly approved (by the Commission on New Minerals, Nomenclature and Classification of the International Mineralogical Association, IMA–CNMNC) nomenclature for the magnetoplumbite group (decision 95–SM/20, Miyawaki et al., Reference Miyawaki, Hatert, Pasero and Mills2020). It should be noted that in this context, we use the commonly accepted formulae of mineral species; the exactness of some of them might be questioned, and a future revision based of reinvestigation of type specimens is desirable.
Table 1. The presently valid magnetoplumbite-group minerals.

* Here renamed chihuahuaite
Crystal structure
Many detailed studies of the crystal structure exist (e.g. Obradors et al., Reference Obradors, Collomb, Pernet, Samaras and Joubert1985; Utsunomiya et al., Reference Utsunomiya, Tanaka, Morikawa, Marumo and Kojima1988; Moore et al., Reference Moore, Sen Gupta and Le Page1989; Wagner Reference Wagner1998). It is based on an essentially closest-packed arrangement of oxygen (O) and A atoms, with B metals occupying voids. One fundamental building block, S, forms a CCP two-layer sequence, ⋅cc⋅. A fraction of the interstitial sites is occupied by metal atoms in the same fashion as in the spinel structure, which gives an overall composition {B 6O8}2+ of the block. A different block, denoted R, is built up of a three-layer HCP sequence, ·hhh·. A quarter of the O atoms of the intermediate h layer is replaced by a large cation A (usually Ba2+, Pb2+, Ca2+ or K+ in minerals). Taking the interstitial B atoms into consideration, R is equal to {AB 6O11}2– in composition. By stacking of the blocks along the hexagonal c axis in the sequence ⋅RSR*S*⋅, with a repeat of 22–23 Å, the magnetoplumbite unit cell with Z = 2 is obtained (Fig. 1). Starred blocks are rotated 180° in accordance with the space-group symmetry of the crystal structure, P63/mmc. The a unit-cell dimension is ~5.6 Å (= 4 × the radius of O2–).
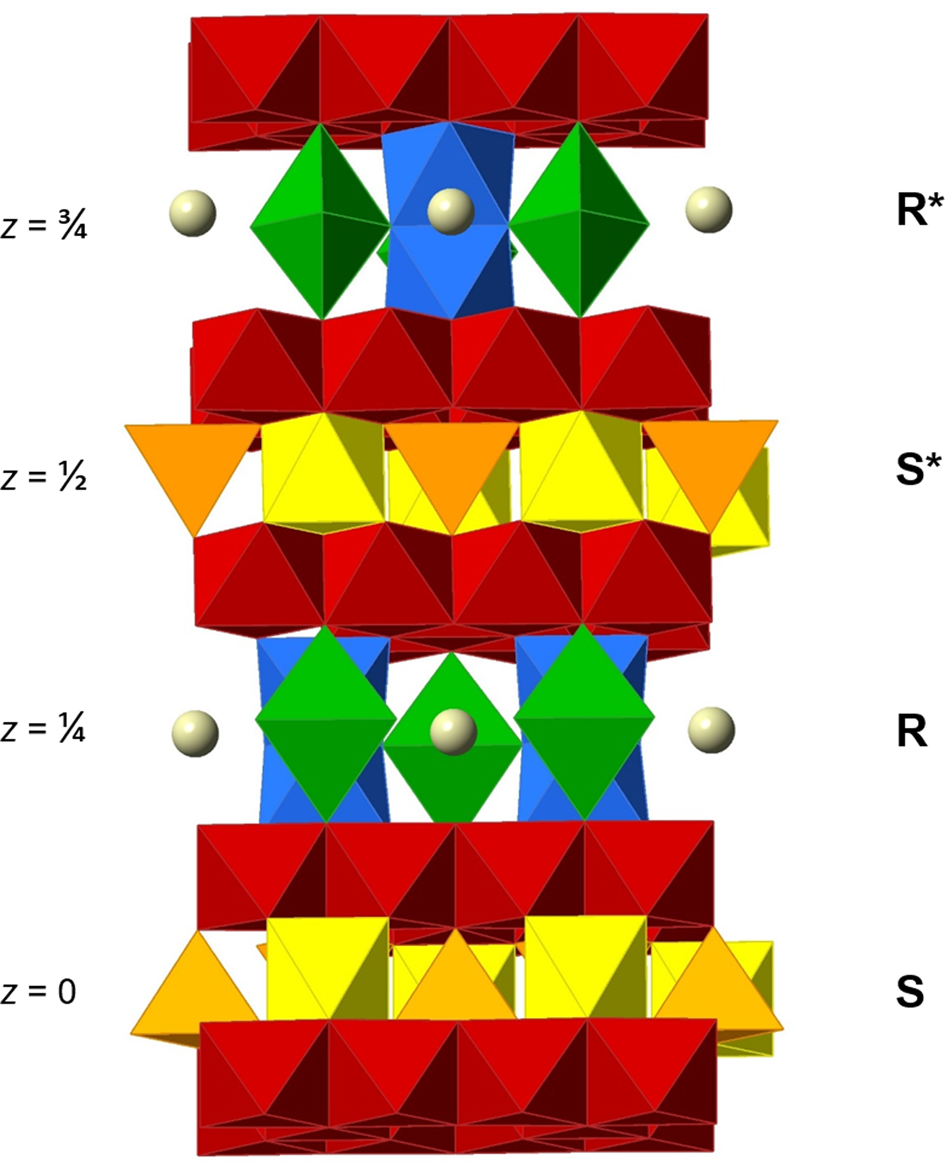
Fig. 1. Polyhedral representation of the ideal magnetoplumbite-type structure viewed approximately along [310]. The M1 octahedra (yellow) and the M3 tetrahedra (orange) are in the central section of the S block. The trigonal bipyramidal M2 positions in (green), face-sharing M4 octahedra (blue) and the large A atoms (grey spheres) belong to the central part of the R block. Layers of edge-sharing M5 octahedra (red) are sandwiched between the cores of blocks.
In the structure, the large A cation is ideally 12-coordinated to O, forming a triangular orthobicupola, at (⅔, ⅓, ¼). The interstitial B atoms occupy five unique sites with designations M1–M5. (Table 2). The five-fold coordinated M2 atom, ideally located at the centre of a trigonal bipyramid (2b), is in reality slightly displaced (split) into two statistically half-occupied, pseudotetrahedral 4e sites (Obradors et al., Reference Obradors, Collomb, Pernet, Samaras and Joubert1985). This kind of disorder is dynamic in most situations, i.e. a rapid diffusion of the metal atom takes place through the mirror plane of the bipyramid (Kimura et al., Reference Kimura, Ohgaki, Tanaka, Morikawa and Marumo1990; Kreisel et al., Reference Kreisel, Lucazeau and Vincent1998; Du and Stebbins, Reference Du and Stebbins2004; Krzątała et al., Reference Krzątała, Panikorovskii, Galuskina and Galuskin2018). The M4 coordination polyhedra are trigonally distorted octahedra that occur in pairs sharing a common face in a hematite-like arrangement, i.e. forming B 2O9 dimers. The total unit-cell contents for an AB 12O19 compound can thus be expressed as A 2[{6}(M1)2 {5}(M2)2 {4}(M3)4 {6}(M4)4 {6}(M5)12]Σ24O38.
Table 2. Properties of crystallographic sites for A and B metal atoms in magnetoplumbite-group minerals.

CN – coordination number
The magnetic structure of magnetoplumbite can be described by the Néel model of ferrimagnetism. The spin orientation of Fe3+ at each site (Table 2) is a result of superexchange interaction through the O2– ions. As the cation has a spin-only magnetic moment of 5 μB (Bohr magnetons), the total magnetisation per formula unit would be (6–2–2 + 1+1) × 5 μB = 20 μB at absolute temperature, which is in good agreement with experimental results (Kojima, Reference Kojima and Wohlfarth1982). Magnetoplumbite possesses a large magnetocrystalline anisotropy, which is related to a strong preference of the magnetic moments of the ions to align along c.
β-alumina (diaoyudaoite), ideally Na[Al11]O17, is a structural derivative of magnetoplumbite (Felsche, Reference Felsche1968) and a common solid-state ion conductor and catalyst. The three O3 atoms at 6h (x, –x, ¼) in the middle h layer of the R block have collapsed to a single point 2c (⅓, ⅔, ¼), compensating for the total lower charge of the metal atoms in this compound. Consequently, R encompasses {AB 5O9}2– and does not contain the nominally 5-coordinated M2 site. The mirror planes at z = ¼ and ¾ correspond to the ion conduction layer in β-alumina.
Nomenclature
Name of the group
Prior to this work, the group had not been formally approved by CNMNC. However the term ‘magnetoplumbite group’ is prevalent in the literature. Strunz and Nickel (Reference Strunz and Nickel2001) denominated the oxide subclass 4.CC.45 as the magnetoplumbite group, which included diaoyudaoite, plumboferrite and lindqvistite. In recent editions of Fleischer's Glossary of Mineral Species (Back, Reference Back2018) the ‘plumboferrite group’, covering the same group of minerals (Table 1), has been introduced. It was then in principle used as a synonym of the magnetoplumbite group.
Although plumboferrite has historical precedence over magnetoplumbite (discovered in 1881 and 1925, respectively), there are several good arguments to keep magnetoplumbite in the group name. In chemistry and materials science, the concept of magnetoplumbite (or simply ‘M’) type compounds for substances possessing a certain crystal structure is extremely well established (e.g. Collongues et al., Reference Collongues, Gourier, Kahn-Harari, Lejus, Thery and Vivien1990; Pullar, Reference Pullar2012). It would be misleading if the mineralogical nomenclature deviated from other areas of science. The true interpretation of the composition of plumboferrite, and its close relationship to magnetoplumbite is in fact a relatively late insight (Holtstam et al., Reference Holtstam, Norrestam and Sjödin1995). Furthermore, plumboferrite is atypical in its formula and slightly different in atomic arrangement compared to other members, including positional disorder of Pb atoms and oxygen vacancies (related to 6s 2 lone electron-pair effects of the Pb2+ ion) in the region of z = ¼ that give rise to weak superstructure reflections in X-ray diffraction data. This species is thus not ideal as an archetype for the group as a whole, although the deviations do not support it to be kept outside the group. The present choice agrees with the statement by Mills et al. (Reference Mills, Hatert, Nickel and Ferraris2009): “a group or a supergroup name can be selected contrary to the precedence rule because the name of this group (supergroup) is very firmly established in the literature.”
Consequences
Although the β-alumina-type minerals, presently diaoyudaoite (Shen et al., Reference Shen, Chen, Li, Dong, Huang and Xu1986) and kahlenbergite, K[Al11]O17 (Krüger et al., Reference Krüger, Galuskin, Galuskina, Krüger and Vapnik2019), were included in a previous grouping, they are not part of the present nomenclature because of the requirement of isostructurality.
The mineral name hibonite-(Fe), for Fe[Al12]O19 (Ma, Reference Ma2010), does not fit well in this scheme as it does not belong to the same subgroup as the parent mineral, hibonite. In addition, suffixes tend to make nomenclature unnecessarily complex. Hibonite-(Fe) is thus assigned a new root name, ‘chihuahuaite’, after the state (estado) of Mexico where Allende, the holotype host meteorite fell in 1969 (King et al., Reference King, Richardson, Schonfeld and Eldridge1969). Levison modifiers may, however, be used if rare earth element (REE) dominant species are to be approved (with new root names).
Lindqvistite, Pb[Fe16Pb(Mn,Mg)]O27, is a related mineral (Holtstam and Norrestam, Reference Holtstam and Norrestam1993). It has the block stacking sequence ⋅RSSR*S*S*⋅ and thus a different topology than magnetoplumbite. Lindqvistite is consequently not counted as a member of the magnetoplumbite group. Galuskin et al. (Reference Galuskin, Galuskina, Widmer and Armbruster2018) have reported closely related Ba- and K-dominant ferrites from Jabel Harmun, West Bank, Palestinian Territories. Further discoveries could motivate the creation of a supergroup, covering different stacking themes among naturally occurring ferrites.
Subdivision
The nomenclature is devised to be simple and flexible at the same time. The group is divided into subgroups based on composition, specifically the dominant A-type cation (Table 3). The rationale for this scheme is that variations in A atom composition tend to be less complex compared to that of B atoms, and information on the precise stoichiometry, including any structural vacancies at cation or anion sites that might be present, is not necessary to determine the position at the highest hierarchal level in the group.
Table 3. Classification of the magnetoplumbite group.

Definition of species
Individual species of the magnetoplumbite group are further defined from their composition and distribution of cations over the B-type positions (Table 4). Monovalent, divalent, tetravalent and pentavalent cations are incorporated in the magnetoplumbite structure by charge-coupled substitutions of A 2+ or B 3+ ions (Table 5). It is evident that a large number of theoretically possible combinations of cation arrangements exist. However, studies on both minerals and synthetic materials show that most cations exhibit preferential ordering depending on their ionic size, charge and electronic configuration (Grey et al., Reference Grey, Madsen and Haggerty1987; Wagner and O'Keefe, Reference Wagner and O'Keeffe1988; Xie and Cormack, Reference Xie and Cormack1990; Bermanec et al., Reference Bermanec, Holtstam, Sturman, Criddle, Back and Scavnicar1996; Holtstam, Reference Holtstam1996; Nagashima et al., Reference Nagashima, Armbruster and Hainschwang2010). An important trend observed is that divalent B-type ions strongly prefer the tetrahedrally coordinated M3 sites (Batlle et al., Reference Batlle, Obradors, Rodriguez-Carvajal, Pernet, Cabañas and Vallet1991), whereas highly charged species, like Ti4+, Mn4+ and Sb5+, become enriched in the M4 octahedra (Doyle et al., Reference Doyle, Schofield, Berry, Walker and Knight2014; Nemrava et al., Reference Nemrava, Vinnik, Hu, Valldor, Kuo, Zherebtsov, Gudkova, Chen, Tjeng and Niewa2017). For compositions with a high degree of replacement of trivalent ions, divalent species also become concentrated at octahedrally coordinated sites, preferentially M5 (Cabañas et al., Reference Cabañas, Gonzlez-Calbet, Rodríguez-Carvajal and Vallet-Regi1994). Some trivalent d cations (Cr3+ and Mn3+) are ordered at the distorted M5 octahedra (e.g. Katlakunta et al., Reference Katlakunta, Meena, Srinath, Bououdina, Sandhya and Praveena2015; Shlyk et al., Reference Shlyk, Vinnik, Zherebtsov, Hu, Kuo, Chang, Lin, Yang, Semisalovaeh, Perov and Langer2015; Nemrava et al., Reference Nemrava, Vinnik, Hu, Valldor, Kuo, Zherebtsov, Gudkova, Chen, Tjeng and Niewa2017). This behaviour is explained largely by crystal-field effects. The Fe3+ cation, in cases when diluted in the compound and less abundant among B positions, e.g. in hibonite, is accumulated at M2 and M3 (Holtstam, Reference Holtstam1996; Medina and Subramanian, Reference Medina, Li and Subramanian2017). Al3+ in turn, when competing with other trivalent species, tends to be concentrated at M1 (Bermanec et al., Reference Bermanec, Holtstam, Sturman, Criddle, Back and Scavnicar1996), with the smallest octahedral volume.
Table 4. Major components at the cation sites of magnetoplumbite-group minerals. Species-defining elements are given in bold.

Table 5. Significant types of substitution in the magnetoplumbite group.

* Describes the relation between magnetoplumbite and plumboferrite, and explains non-stoichiometry in some synthetic magnetoplumbite samples (Holtstam, Reference Holtstam2003).
New or unaccredited mineral compositions
In the literature, analytical data are available that suggest the existence of new, yet officially unrecognised members of the group. Titanium-rich analogues of yimengite and hawthorneite (~5 Ti atoms per formula unit) were analysed by Lu and Chou (Reference Lu and Chou1994). Lu et al. (Reference Lu, Liu and Xiao2007) have described a “Ca analogue to yimengite” or rather a Ca analogue to hawthorneite, which would fit into the hibonite subgroup. Rezvukhin et al. (Reference Rezvukhin, Alifirova, Korsakov and Golovin2019) recently found yimengite with high Al (>1 atom per formula unit) contents. Sandiford and Santosh (Reference Sandiford and Santosh1991) described zoned ‘hibonite’ grains with REE-rich cores (ΣREE > 0.6 atoms per formula unit). Holtstam (Reference Holtstam1994) reported a Ti-rich magnetoplumbite sample for which Ti > Fe3+ at M4 could be inferred (a possible Pb analogue to batiferrite). A Mn3+-analogue to plumboferrite was detected by Chukanov et al. (Reference Chukanov, Aksenov, Jančev, Pekov, Göttlicher, Polekhovsky, Rusakov, Nelyubina and Van2016). Furthermore, Chukanov et al. (Reference Chukanov, Vorobei, Ermolaeva, Varlamov, Plechov, Jančev and Bovkun2019) recently published analyses of a Ba-dominant analogue to nežilovite and of an Al analogue to yimengite.
From a vast amount of studies of synthetic compounds, it can be speculated that many new natural members exist with, for example: A = Sr2+, REE (Ce3+, La3+ etc.), Mg2+, Rb+, Cs+ or Ag+ along with enrichment in the B positions (non-exhaustive list) of: Si4+, Sc3+, Ti3+, V2+, V3+, V4+, Co2+, Ni2+, Cu2+, Ga3+, Ge4+, Zr4+, Nb5+, In3+, Sn4+, Te4+, Ta5+ or Bi3+ (e.g. Coutellier et al., Reference Coutellier, Ferrand, Daval, Grange and Joubert1984; Morgan and Miles, Reference Morgan and Miles1986; Li et al., Reference Li, Medinam, Stalick, Sleight and Subramanian2016). The range of possible cation valences seem to be limited to 1–3 for A and 2–5 for B sites, which has implications when casting formulae of uncharacterised members of the group. Particular caution is needed for samples containing some of the divalent ionic species, as Fe2+, Mg2+ and Pb2+ have been shown to enter both kinds of sites. Substitutions at anion sites seem to be limited for this structure type.
Acknowledgements
Constructive comments on the original nomenclature proposal by members of the Commission on New Minerals, Nomenclature and Classification (CNMNC), and on the manuscript by two reviewers, are appreciated.







 Open access
Open access