


Long-term high fructose consumption is known to induce hyperglycaemia, hypertriacylglycerolaemia, insulin resistance and hypertension in the rat(Reference Hwang, Ho and Hoffman1). The potential mechanisms proposed by which hyperinsulinaemia causes hypertension in this model(Reference DeFronzo and Ferrannini2) has been Na retention, sympathetic nerve activation and vascular smooth muscle proliferation. It has also been reported that elevated sympathetic nerve activity associated with insulin resistance may contribute to the onset and maintenance of cardiovascular and renal complications(Reference Masuo, Rakugi and Ogihara3). Furthermore, insulin resistance in rat models of the metabolic syndrome has been suggested to be closely associated with abnormal pressure natriuresis and hypertension(Reference Fujiwara, Hayashi and Matsuda4), but changes in vascular reactivity are also considered to be a contributory factor to the hypertensive state(Reference Yoshida, Ohyanagi and Iwasaki5).
The functionally relevant adrenoceptors at the renal vasculature and epithelial cells of the nephron are of the α1-adrenoceptor subtype(Reference Johns and Manitius6). It has been reported that the density of α1-adrenoceptors is highest in the cortex and progressively decreases from the cortex to the papilla(Reference Feng, Pettinger and Abel7). The effects of insulin resistance on α1-adrenoceptor-mediated vasoconstriction of resistance vessels, and therefore blood flow to the tissues, have not been fully elucidated. It has been reported that α1-adrenoceptor-mediated vasoconstriction was enhanced in large arterial vessels(Reference Tuck8) as well as in small arterioles(Reference Yoshida, Ohyanagi and Iwasaki5) in insulin-resistant type 2 diabetic rats. This augmentation was suggested as being secondary to impaired insulin-dependent vasodilatation in these rats(Reference Feldman and Bierbrier9, Reference Baron10). Renal α-adrenoceptors are known to be the major functional type in the rat renal vasculature and play a pivotal role in mediating the response to sympathetic stimulation(Reference Salomonsson, Brannstrom and Arendshorst11). It has been reported that both α1A- and α1D-adrenoceptor subtypes are expressed in the rat renal artery(Reference Salomonsson, Brannstrom and Arendshorst11, Reference Villalobos-Molina, Lopez-Guerrero and Ibarra12) and that the adrenergically induced renal vasoconstriction is mediated predominantly by α1A- and α1D-adrenoceptors in normal and hypertensive streptozotocin-induced diabetic Sprague–Dawley rats(Reference Sattar, Abdullah and Khan13, Reference Armenia, Munavvar and Abdullah14).
In an in vivo study, we have shown that α1-adrenoceptor-mediated cortical vasoconstriction is attenuated by an insulin-resistant state. Together, these previous reports have suggested that in an insulin-resistant state, adrenergically mediated renal vasoconstriction was depressed. The present investigation aimed to provide further evidence for this view by exploring another model of insulin resistance, the fructose-fed rat (FFR). The hypothesis explored was that the α1-adrenoceptor-mediated renal vasoconstrictor responses were shifted to the α1D-adrenoceptor subtype in the fructose-fed model of insulin resistance.
Materials and methods
Animals
A total of thirty-two male Sprague–Dawley rats (158–195 g) were obtained from the Central Animal Facility at Universiti Sains Malaysia, Penang, Malaysia. The rats were allowed to acclimatise to the new environment (controlled conditions of temperature and humidity and on a 12 h light–12 h dark cycle) for at least 5 d and were permitted free access to tap water and standard rodent chow (Gold Coin Sdn. Bhd., Penang, Malaysia). Thereafter, the rats were assigned randomly into two groups (n 16), namely control (C), which received a standard rodent chow and tap water ad libitum, and FFR, which were fed a standard rodent chow and fructose was administered as a 20 % solution (prepared freshly every day) in drinking water ad libitum. Each of the aforementioned groups were subdivided into two subgroups (n 8), one of the subgroups entered into the study (protocol 1) for 8 weeks while the other entered the acute haemodynamic study (protocol 2). All the procedures and experiments were approved by the Ethics Committee of Universiti Sains Malaysia and a written agreement was obtained (approval letter ref. USM/PPSF/50 (96) Jld.1).
Protocol 1
The rats were subjected to weekly metabolic studies over the 8 weeks during which several measurements were performed. Body weight, fluid intake, food intake, non-fasting and fasting blood glucose levels and plasma levels of TAG, cholesterol and albumin of each rat were measured on a weekly basis for 8 weeks. Plasma insulin levels were measured at the end of the treatment period. Food and fluid intakes for each rat were measured by subtracting the amounts remaining in the metabolic cages after 24 h from the initial quantities provided. Blood samples (300 μl) were collected from the tail weekly throughout the study period in non-fed and fed states. Plasma insulin level was measured using a quantitative Ultra-Sensitive Rat Insulin ELISA kit (Crystal Chem, Inc., Downers Grove, IL, USA). The reactivity for rat insulin is 100 % according to the manufacturer. Plasma TAG, cholesterol and albumin were measured using a biochemical auto analyser (ChemWell®; Awarness Technology, Inc., Palm City, FL, USA).
Intravenous insulin glucose tolerance test
This test was performed to examine insulin sensitivity in rat. An intravenous insulin glucose tolerance test (IVIGTT) was carried out as described previously with minor modifications(Reference Viswanad, Srinivasan and Kaul15). After the 8-week feeding period, the overnight fasted rats (12 h) were anaesthetised with 60 mg/kg (intraperitoneal) sodium pentobarbitone (Nembutal®; CEVA, Libourne, France). Following tracheostomy, the right carotid artery was cannulated (PP50 Portex, Kent, UK). The left jugular vein was cannulated to infuse glucose, insulin and anaesthetic when required. Following 30 min of stabilisation, the rats were successively injected with glucose (0·7 g/kg) and insulin (0·175 U/kg) into the jugular vein. Blood samples (20 μl) were then withdrawn from the carotid artery at approximately 0 (before glucose), 2, 4, 6, 8, 10, 20 and 30 min after insulin injection for estimation of glucose levels using the ACCU-CHEK advantage blood glucose monitoring system (Roche Diagnostics Corporation, Indianapolis, IN, USA). An equivalent volume of saline (0·9 %, w/v) was injected into the jugular vein following each sampling to prevent any change in the central blood compartment.
Blood pressure measurements
Systolic blood pressure (SBP) measurement was performed using the tail-cuff method in conscious animals as reported previously(Reference Navarro-Cid, Maeso and Perez-Vizcaino16, Reference Huang, Boini and Friedrich17). The rats underwent a 3 d training session before commencing the actual recording. SBP was measured once the rats were considered to be trained and not susceptible to stress from the tail-cuff procedure. Blood pressure readings were taken from each rat in a calm and dark room, and an average value of ten consecutive SBP measurements was calculated. Readings that deviated by not more than 5 mmHg were selected.
Protocol 2
Animals in this part of the study underwent the acute renal vasoconstrictor response study after 8 weeks of feeding either as a control or a FFR.
Animal surgical preparation
Renal vasoconstrictor responses
In an in vivo renal vasoconstrictor response experiment which was adapted from previous studies(Reference Hye Khan, Sattar and Abdullah18–Reference Abdul Sattar and Johns20), the rats were fasted overnight before being anaesthetised with 60 mg/kg intraperitoneal sodium pentobarbitone (Nembutal®; CEVA). Thereafter, the trachea was exposed through a midline incision and cannulated (PE250, Portex). Then, the left carotid artery was cannulated (PE50, Portex) and attached to a fluid-filled pressure transducer (model P23 ID Gould; Statham Instruments, Nottingham, UK) connected to a computerised data acquisition system (PowerLab®; ADInstruments, Sydney, Australia) for continuous monitoring of mean arterial blood pressure (MAP) and heart rate. The left jugular vein was cannulated (PE50, Portex) to infuse anaesthetic when needed. The aorta and the left kidney were exposed via a midline abdominal incision. A laser Doppler probe (OxyFlo® Probe; Oxford Optronix Limited, Oxford, UK) positioned in the outermost layer of the kidney, and connected to a laser Doppler flowmeter (ADInstruments, Sydney, Australia), was used to measure renal cortical blood flow (CBF) continuously throughout the experiment(Reference Roman, Mattson and Cowley21). The CBF data were further analysed using a computerised data acquisition and analysing system (PowerLab®; ADInstruments). A cannula (PE50, Portex) was inserted via the left common iliac artery, such that its tip lay close to the entrance of the renal artery to administer noradrenaline (NA), phenylephrine (PE), methoxamine (ME) and angiotensin II (Ang II) into the renal artery. The cannula was kept patent by infusing saline at a rate of 6 ml/kg per h. The iliac artery cannula was attached to a second pressure transducer (model P23 ID Gould; Statham Instruments) linked to a computerised data acquisition system (PowerLab®; ADInstruments) for baseline measurements of renal arterial pressure. The urinary bladder was cannulated to allow free passage of urine gravimetrically. On completion of the surgery, a stabilisation period of 1 h was allowed before entering into the experimental protocol.
Renal vasoconstrictor response experimental protocol
The renal vasoconstrictor experiments were performed in three phases as mentioned previously(Reference Armenia, Munavvar and Abdullah14, Reference Khan, Sattar and Abdullah22, Reference Khan, Sattar and Abdullah23).
After the stabilisation period, baseline measurements of MAP, heart rate, renal arterial pressure and CBF were recorded for 3 min and thereafter dose–response curves to NA, PE, ME and Ang II were generated. This involved assessing the renal CBF responses to bolus doses of NA at 25, 50, 100 and 200 ng, PE at 0·25, 0·50, 1 and 2 μg, ME at 0·5, 1, 2 and 4 μg and Ang II at 2·5, 5, 10 and 20 ng.
During the first phase, the rats received vehicle (normal saline, NaCl 9 g/l) at 6 ml/kg per h along with the adrenergic agonists and Ang II into the renal artery line. In the second phase, a low bolus dose of BMY7378 (100 μg/kg) was injected slowly over 30 s followed by a continuous infusion of BMY7378 (25 μg/kg per h) into the renal artery line, and 15 min later, the second set of renal vasoconstrictor responses to NA, PE, ME and Ang II was performed. In the third and last phase, a high bolus dose of BMY7378 (200 μg/kg) was injected slowly over 30 s followed by a continuous infusion of BMY7378 (50 μg/kg per h). Upon reaching the steady state, 15 min later, the same procedure used in the first and second phases was followed. The administration of agonists was carried out twice in an ascending order followed by descending order of doses. The doses of the agonists and antagonist were adapted from previous work in this laboratory(Reference Khan, Sattar and Abdullah23–Reference Kazi, Munavvar and Abdullah25) and aimed to produce a local action without any significant effect on the systemic blood pressure.
Vasoactive agents
NA (Sanofi Winthrop, Surrey, UK), PE (Knoll, Nottingham, UK), ME (Wellcome, London, UK) and Ang II (CIBA-GEIGY, Basel, Switzerland) were used in the renal vasoconstrictor experiment. All drugs were prepared as stock solutions in normal saline on the day of the experiment and stored at +4°C.
Antagonist used
BMY7378 (8-(2-[4-(2-methoxyphenyl)-1-piperazinyl) ethyl]-8-azaspiro (4·5) decane-7,9-dione dihydrochloride; Research Biochemicals International, Natick, MA, USA) is a selective antagonist for the α1D-adrenoceptor subtype(Reference Armenia, Munavvar and Abdullah14, Reference Arevalo-Leon, Gallardo-Ortiz and Urquiza-Marin26, Reference Goetz, King and Ward27).
Statistical analysis
The maximum CBF decrease following each injection of the agonists was determined off-line using the software (LabChart 6; ADInstruments, Sydney, Australia) and is expressed as a percentage of the baseline value. Baseline CBF and MAP were determined from the chart immediately before each agonist injection. The vasoconstrictor responses caused by Ang II and adrenergic agonists were taken as the average values caused by each dose of the agonists administered in ascending and descending orders. The mean values for every phase (Figs. 2 and 3) are the overall mean calculated for all doses of each agonist and compared between high- and low-dose antagonist phases, and the saline phase. All data are expressed as means with their standard errors. The statistical analysis of the dose–response data utilised two-way ANOVA followed by the Bonferroni post hoc test using the statistical package Superanova (Abacus Inc., Sunnyvale, CA, USA). However, the analysis of body weight, fluid intake, food intake, non-fasting and fasting blood glucose levels and plasma TAG, cholesterol and albumin throughout the 8-week feeding period, and baseline haemodynamic parameters measured during the acute experiment, were analysed using repeated-measures one-way ANOVA followed by the Bonferroni post hoc test for the differences between weeks 1–8 and week 0 or low- and high-dose phases and the saline phase. The unpaired t test was used to detect the differences between the C and FFR groups. The differences between the means were considered significant at the 5 % level.
Results
The effect of fructose feeding on metabolic and haemodynamic parameters
A comparison of the metabolic data between the FFR and C rats is shown in Table 1. The IVIGTT was performed at the end of the 8-week treatment period, and showed that FFR had less tolerance to intravenous glucose load (higher area under the curve, P < 0·0001) compared with the C group (113 (se 6) v. 188 (se 9), n 7). Moreover, FFR developed a higher SBP as early as 1 week compared with the C group and remained consistently higher until the end of the study; however, it only achieved significance at weeks 4, 6 and 8 (all P < 0·05; Table 1). Food intake of FFR throughout the study was significantly lower (P < 0·01) than the C group (Fig. 1). In addition, significant increases in body weights compared with baseline values in both C rats and FFR started after 3–4 weeks and continued until the end of the treatment period (Fig. 1). FFR at week 8 of the feeding period had higher values of body weight (P = 0·0006), fasting blood glucose (P = 0·0005), non-fasting blood glucose (P = 0·0008), plasma TAG (P = 0·005) and plasma cholesterol (P = 0·01; Fig. 1 and Table 1). The plasma insulin level at the end of the 8-week feeding period in the FFR was higher than the C group (2·83 (se 0·2) v. 1·89 (se 0·2); P = 0·02, n 5). However, no significant difference was observed in fluid intake (P = 0·56) and plasma albumin (P = 0·82) between the FFR and C rats at the end of the 8-week feeding period (Fig. 1 and Table 1).
Table 1 Metabolic and haemodynamic parameters during the 8-week feeding period in control (C) and fructose-fed rats (FFR)
(Mean values with their standard errors)

SBP, systolic blood pressure; FBG, fasting blood glucose; NFBG, non-fasting blood glucose; Ch, cholesterol; Alb, albumin.
* Mean values were significantly different between the FFR and C groups during that particular week (P < 0·05).
† Mean values were significantly different between every week and week 0 in C rats (P < 0·05).

Fig. 1 Food intake, fluid intake and body weight of control (C, –○–) and fructose-fed rats (FFR, –●–) during the 8-week feeding period. Values are means, with their standard errors represented by vertical bars (n 8). * Mean values were significantly different between every week and week 0 in the C group (P < 0·05). ‡ Mean values were significantly different between the FFR and C rats during that particular week (P < 0·05). † Mean values were significantly different between every week and week 0 in the FFR group (P < 0·05).
Baseline haemodynamic parameters during the acute vasoconstrictor experiment
Baseline values of SBP, diastolic blood pressure, MAP and renal arterial pressure measured during the saline phase of the acute experiment were significantly higher in the FFR (all P < 0·05) than the C group; however, baseline CBF and heart rate of the FFR were similar to the C group (Table 2). In addition, in the second (low-dose) and third (high-dose) BMY7378 phases of the acute experiment, there were no significant alterations in baseline values of SBP, diastolic blood pressure, MAP and renal arterial pressure in the FFR or C group compared with the saline phase (Table 2). The baseline heart rate values of the third phase in the FFR was significantly (P = 0·003) lower than the saline phase (Table 2) while the baseline CBF values in the low- or high-dose antagonist phases in the FFR or C group, although lower, were not significantly different from the saline phase (Table 2).
Table 2 Systolic blood pressure (SBP), diastolic blood pressure (DBP), mean arterial pressure (MAP), heart rate (HR), renal arterial pressure (RAP) and cortical blood flow (CBF) values measured during the three-phase acute renal vasoconstrictor experiment in control (C) and fructose-fed rats (FFR)
(Mean values with their standard errors)

bpm, Beats per minute; bpu, blood perfusion unit.
† Mean values were significantly different between the low- or high-dose phases and the saline phase in the FFR group (P < 0·05).
‡ Mean values were significantly different between the FFR and C groups during that particular phase (P < 0·05).
Renal cortical vasoconstrictor response
Adrenergic agonists
Exogenously administered NA, PE and ME resulted in dose-dependent renal cortical vasoconstrictions in the saline phase of both C rats and FFR (Figs. 2 and 3) but the overall responses were significantly lower (all P < 0·05) in the FFR compared with the C group (FFR: NA, 16 (se 3) %; PE, 22 (se 3) %; ME; 9 (se 1) % v. C: NA, 32 (se 4) %; PE, 43 (se 4) %; ME, 26 (se 4) %; P = 0·0002, P = 0·0002, P = 0·02), respectively (Figs. 2–4). In the later subsequent phases of the experiment, the renal cortical vasoconstrictions induced by NA, PE and ME were attenuated by both the low dose of BMY7378 (low-dose BMY phase: NA, 22 (se 3) %; PE, 33 (se 4) %; ME, 21 (se 3) %; P = 0·007, P = 0·02, P = 0·15), respectively, but to a greater extent by the high dose of BMY7378 in C rats relative to their corresponding saline phase (high-dose BMY phase: NA, 22 (se 3) %; PE, 29 (se 4) %; ME, 15 (se 2) %; P = 0·005, P = 0·001, P = 0·002), respectively (Figs 2 and 3). In the FFR, the NA-, PE- and ME-induced renal vasoconstrictions were enhanced significantly (all P < 0·05) by the high dose of BMY7378 (high-dose BMY phase: NA, 29 (se 3) %; PE, 34 (se 4) %; ME, 25 (se 4) % v. saline phase: NA, 16 (se 3) %; PE, 22 (se 3) %; ME; 9 (se 1) %; P = 0·0001, P = 0·004, P = 0·0001), respectively (Figs 2 and 3). The low dose of BMY7378 in the FFR produced no significant alteration in the magnitude of the renal cortical vasoconstriction responses to the adrenergic agonists compared with the saline phase (low-dose BMY phase: NA, 17 (se 2) %; PE, 22 (se 3) %; ME, 9 (se 2) %; P = 0·66, P = 0·98, P = 0·96), respectively (Figs 2 and 3). In addition, the magnitude of the BMY7378-induced change in the renal cortical vasoconstriction due to NA, PE and ME was significantly higher during the high-dose phase compared with the low-dose phase in the FFR (P = 0·0002, P = 0·004, P = 0·0001), respectively, but not in C rats (P = 0·88, P = 0·39, P = 0·09) (Figs. 2 and 3).
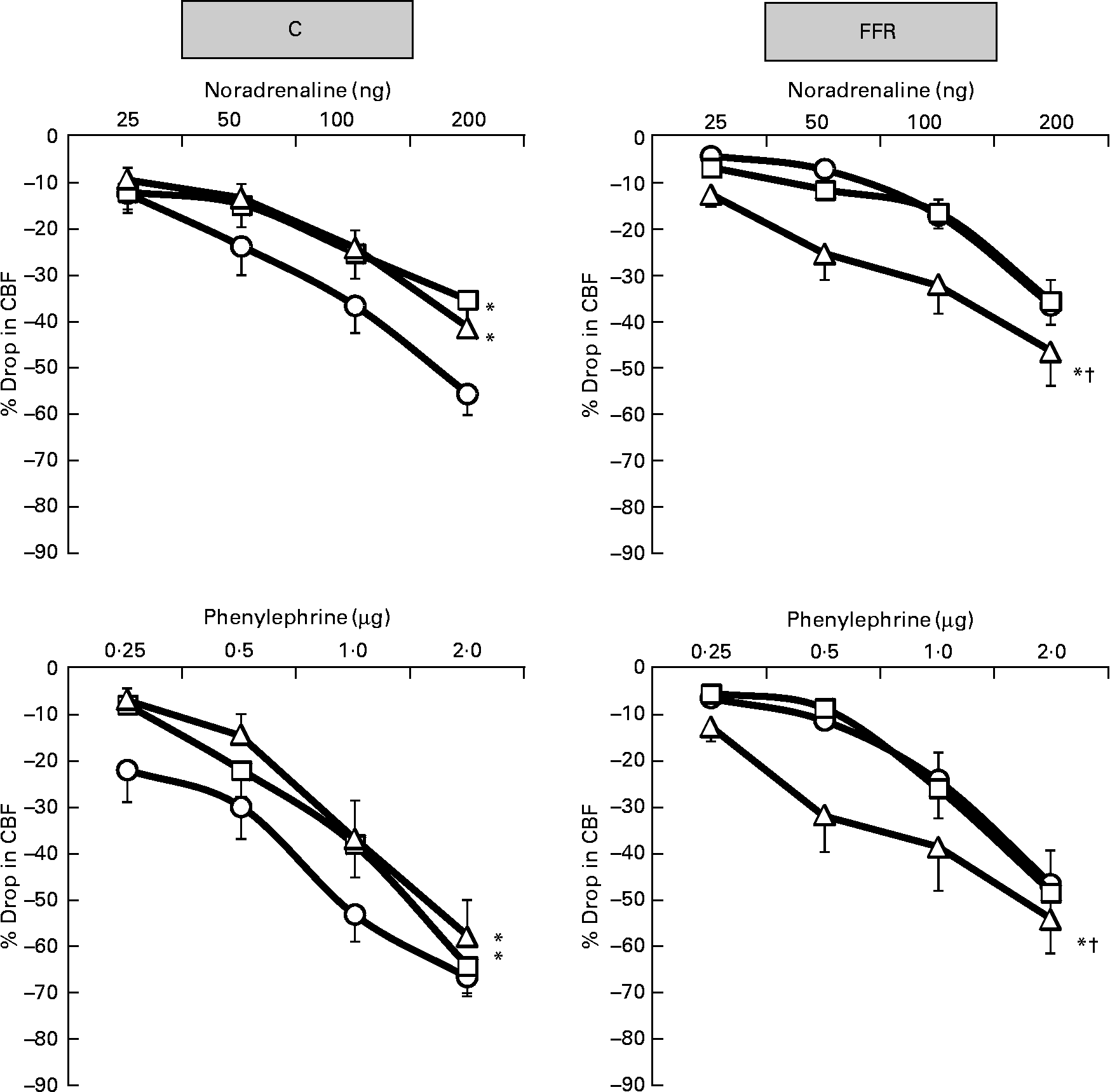
Fig. 2 Dose–response curve of the renal vasoconstrictor responses to graded doses of noradrenaline and phenylephrine in control (C) and fructose-fed rats (FFR) during the saline phase (–○–), low dose of BMY7378 (–□–) and high dose of BMY7378 (–△–). Values are means, with their standard errors represented by vertical bars (n 8). The significance is between the overall mean of responses due to four doses of agonist during each phase and compared with the saline phase. * Mean values were significantly different from those of the saline phase (P < 0·05). † Mean values were significantly different between the low- and high-dose BMY7378 phases (P < 0·05). CBF, cortical blood flow.
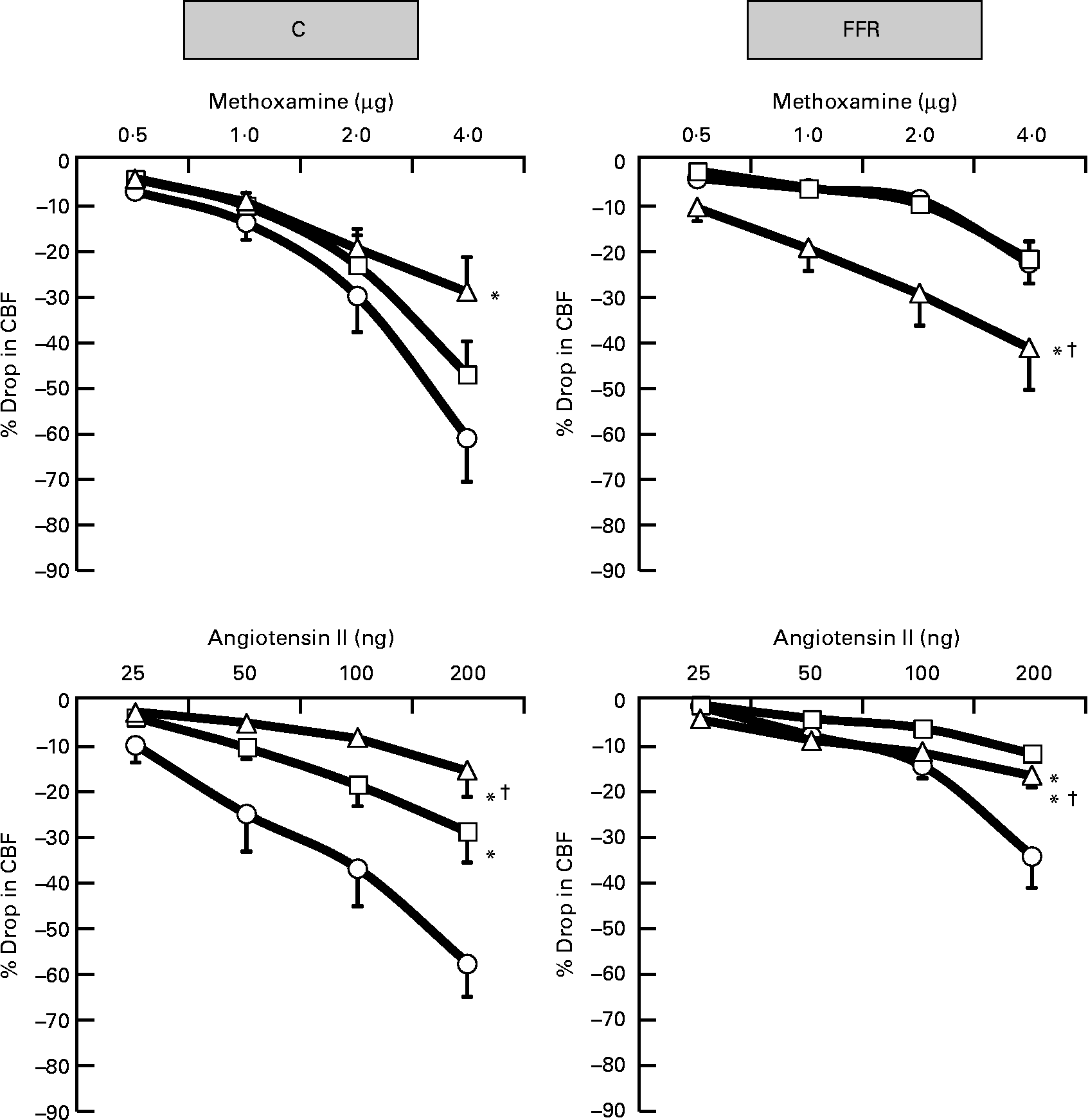
Fig. 3 Dose–response curve of the renal vasoconstrictor responses to graded doses of methoxamine and angiotensin II in control (C) and fructose-fed rats (FFR) during the saline phase (–○–), low dose of BMY7378 (–□–) and high dose of BMY7378 (–△–). Values are means, with their standard errors represented by vertical bars (n 8). The significance is between the overall mean of responses due to four doses of agonist during each phase and compared with the saline phase. * Mean values were significantly different from those of the saline phase (P < 0·05). † Mean values were significantly different between the low- and high-dose BMY7378 phases (P < 0·05). CBF, cortical blood flow.

Fig. 4 Effect of 8 weeks of fructose feeding on the renal vascular responses to adrenergic agonists and angiotensin II (Ang II). Values are means from saline phases of the control (C, ![]() ) and fructose-fed rats (FFR,
) and fructose-fed rats (FFR, ![]() ), with their standard errors represented by vertical bars (n 8). * Mean values were significantly different between the FFR and C groups (P < 0·05). CBF, cortical blood flow; NA, noradrenaline; PE, phenylephrine; ME, methoxamine.
), with their standard errors represented by vertical bars (n 8). * Mean values were significantly different between the FFR and C groups (P < 0·05). CBF, cortical blood flow; NA, noradrenaline; PE, phenylephrine; ME, methoxamine.
Angiotensin II
Ang II produced dose-related decreases in CBF in the saline phase of both C and FFR groups (Fig. 3). The Ang II-induced renal cortical vasoconstrictions in the FFR were significantly lower compared with C rats (FFR: 15 (se 3) % v. C: 33 (se 4) %, P < 0·0001; Fig. 4). In the presence of the low and high doses of BMY7378 in both FFR and C groups, the reduction in CBF due to Ang II was smaller (P < 0·05) compared with the saline phase (FFR – low-dose BMY phase: 6 (se 0·7) % and high-dose BMY phase: 10 (se 1) % v. saline phase: 15 (se 3) %; P = 0·0001, P = 0·004); C – low-dose BMY phase: 16 (se 3) % and high-dose BMY phase: 8 (se 2) % v. saline phase: 33 (se 4) %; P = 0·0001, P = 0·0001; Fig. 3). Moreover, the magnitude of the BMY7378 attenuation of the renal cortical vasoconstriction due to Ang II was significantly higher during the high-dose phase compared with the low-dose phase in C rats, while it was significantly lower in FFR (P = 0·03, P = 0·007), respectively (Fig. 3).
Discussion
The present study investigated the effect of a chronic 8-week period of high fructose consumption on the role of the α1D-adrenoceptor subtype-mediated renal vasoconstrictor responses. It was evident that the long-term high fructose intake produced an elevation in blood pressure, hypertriacylglycerolaemia, hyperinsulinaemia and, most importantly, renal contractile dysfunction. The mechanisms that triggered the elevation in blood pressure in this model are not fully understood and remain a subject of debate. One of the proposed mechanisms for hypertension associated with insulin resistance is an augmented sympathetic activity(Reference Verma, Bhanot and McNeill28), and an increase in renal Na reabsorption(Reference DeFronzo29). An augmentation in arteriolar vasoconstriction has also been proposed as a cause of elevated blood pressure in insulin resistance(Reference Yoshida, Ohyanagi and Iwasaki5). In addition, diminished peripheral vasodilatory capacity associated with NO depletion has been reported in the FFR(Reference Lin, Tseng and Chang30). Moreover, increased vascular tone and endothelial dysfunction have been proposed to contribute to the development of hypertension and/or insulin resistance(Reference Hsueh and Anderson31–Reference Luscher, Bock and Yang33). The present study showed that 8 weeks of fructose feeding not only caused hyperinsulinaemia and elevated blood pressure but also resulted in hypertriacylglycerolaemia, hypercholesterolaemia, hyperglycaemia and weight gain, and this is well in agreement with previous reports(Reference Hwang, Ho and Hoffman1, Reference Farah, Elased and Chen34–Reference Park, Cesar and Faix37). In addition, the intrarenal administration of low or high dose of BMY 7378 in the present study did not lead to significant changes in the baseline haemodynamic parameters in either group compared with the saline phase.
The present study aimed to investigate in depth the role of different α1-adrenoceptor subtypes and Ang II that contribute to a suppressed reactivity. The pattern of the vasoconstrictor responses obtained in the FFR was similar to that of the C group except that the magnitudes of the renal cortical vasoconstrictor responses induced by all agonists in the FFR were less than in controls. This would be consistent with earlier observations that in rats fed with fructose chronically for 13–18 weeks(Reference Bunnag, Hori and Ormsby38), there was an attenuated sensitivity to exogenous NA both in vivo and in vitro. Furthermore, in a 4-week FFR(Reference Kamide, Rakugi and Higaki39), there was an increase in urinary excretion of NA. It has been argued that this might be due to a compensatory change that follows the enhanced activity of the sympathetic nervous system resulting in desensitisation or down-regulation of the receptors(Reference Sun and Hanig40, Reference Hogikyan and Supiano41). In addition, it has been reported that the insulin-resistant state is related to an enhanced function of Ang II and hyperactivity of tissue AT1-receptors in high FFR(Reference Shinozaki, Ayajiki and Nishio42); however, the vasopressor responses to Ang II in a 4-week FFR tended to be lower than the C group(Reference Iyer and Katovich43). Indeed, we have recently shown that FFR had higher plasma level of Ang II compared with the C group(Reference Abdulla, Sattar and Johns44). Furthermore, Stepp et al. (Reference Stepp, Boesen and Sullivan45) observed no difference in reactivity to Ang II in the renal vasculature after 8 weeks of fructose feeding. Despite these conflicting observations, under the conditions of the present study, there was a blunted renal cortical vasoconstriction to exogenous Ang II and adrenergic agonists, suggesting that high-fructose feeding attenuated the sensitivity of the renal cortical vasculature to the administered agonists. This suggestion is supported by the findings of higher circulating levels of catecholamines and Ang II in the FFR(Reference Tran, Yuen and McNeill46). The argument that we can provide here is that the mentioned studies used different approaches to study the vascular reactivity compared with the present study, with the current study mainly focusing on a renal vasculature-specific approach. In addition, the arterial hyperactivity, which was shown in the aorta(Reference Shinozaki, Ayajiki and Nishio42), may not necessarily reflect the situation at the level of the renal microcirculation where a variety of phenotypes and different haemodynamic forces exist compared with those in the aorta(Reference Stepp, Boesen and Sullivan45). Moreover, in the previous in vivo studies in the rat, the agonists were infused intravenously into the systemic circulation(Reference Bunnag, Hori and Ormsby38, Reference Iyer and Katovich43) and not directly into the renal artery.
It was evident that in normal rats, the renal vasoconstrictor responses elicited by exogenously administered adrenergic agonists were significantly blunted by BMY 7378, which agrees well with previous reports from this laboratory in normal and hypertensive rats(Reference Sattar, Abdullah and Khan13, Reference Armenia, Munavvar and Abdullah14, Reference Hye Khan, Sattar and Abdullah18, Reference Armenia, Sattar and Abdullah19). By contrast, BMY7378 in the present study at both low and high doses significantly enhanced the cortical vasoconstriction in response to adrenergic agonists in the FFR, with the high-dose phase characterised by a greater renal cortical haemodynamic change compared with the low-dose phase. The exact mechanism underlying this type of response is not clear; however, there is a possibility that the BMY7378 antagonism of postsynaptic α1D-adrenoceptors interacts and enhances the sensitivity and contribution of the other α1-adrenoceptor subtypes, which are also involved in the vasoconstrictor responses(Reference Armenia, Munavvar and Abdullah14, Reference Khan, Sattar and Abdullah22). Such an antagonistic type of relationship between α1-adrenoceptor subtypes does exist in the neonatal rat cardiovascular system(Reference Deng, Sculptoreanu and Mulay47). In addition, augmented renal vasoconstrictor responses have been reported in several pathophysiological states(Reference Armenia, Munavvar and Abdullah14, Reference Abdul Sattar and Johns20, Reference Khan, Sattar and Abdullah22). There has been a suggestion that there is a reduction in the ability of the α1D-adrenoceptor subtypes to mediate vasoconstriction when there is a high sympathetic drive. It has been shown that the population of receptors in the blood vessels is influenced by a feedback mechanism that desensitises the α1D-adrenoceptor, as it could easily be phosphorylated(Reference Garcia-Sainz, Vazquez-Cuevas and Romero-Avila48). Therefore, the result is an enhancement in the functional contribution of other α1-adrenoceptor subtype (especially α1A-adrenoceptors) and, indeed, fructose induced hypertension has been associated with an elevated sympathetic activity(Reference Verma, Bhanot and McNeill28). Moreover, this view is supported by the observation that the α1D-adrenoceptor subtypes are phosphorylated in the face of enhanced sympathetic activity(Reference Arevalo-Leon, Gallardo-Ortiz and Urquiza-Marin26, Reference Garcia-Sainz, Vazquez-Prado and del Carmen Medina49). It is therefore possible to conclude that these results support a functional contribution of α1D-adrenoceptors in mediating adrenergically induced renal vasoconstriction in normal rats but not in the FFR.
It has been suggested that α1-adrenoceptors are involved in the process of sequestration and receptor down-regulation(Reference Garcia-Sainz50), which has been reported to be prolonged in pathophysiological states characterised by exaggerated sympathetic activity. In relation to that, down-regulation of certain α1-adrenoceptors has been reported in the renal vasculature of rats with renal impairment(Reference Khan, Sattar and Abdullah22).
In addition, previous studies have reported a possible down-regulation of postsynaptic α1B-adrenoceptors by the adrenergic nerves with increased contribution of other subtypes such as α1A-adrenoceptors in order to maintain the effectiveness of the α1-adrenoceptor neuro-transmission system(Reference Armenia, Munavvar and Abdullah14). Therefore, it is possible that down-regulation of renal α1-adrenoceptor in the present study is responsible for the blunted vascular responses of the FFR group compared with the C group. Interestingly, we have recently shown that there is a higher contribution of α1A-adrenoceptor subtype(Reference Abdulla, Sattar and Johns44) compared with α1B subtype(Reference Abdulla, Sattar and Abdullah51) in the renal vasculature of FFR.
The interaction between AT1 receptors and α1-adrenoceptors in the renal vasculature has been extensively reported in this laboratory and others(Reference Abdulla, Sattar and Abdullah24, Reference Abdulla, Sattar and Khan52, Reference Garcia, Monasterolo and Elias53). Since activation of both receptors initiates common signalling pathways, there will be physiological interactions at both cellular and molecular levels between the AT1 and α-adrenoceptors. Sumners & Raizada(Reference Sumners, Raizada, Raizada, Phillips and Sumners54) reported that a long term NA activation of α1-adrenoceptors at neurons results in the down-regulation of AT1-receptors. On the other hand, Ang II induces transcription and expression of α1-adrenoceptors in the rat vasculature(Reference Hu, Shi and Okazaki55). Interestingly, renal cortical vasoconstriction to Ang II in the present study was significantly blunted following the administration of BMY7378 in both C and FFR groups. Thus, the possibility arises that there is a cross-talk relationship between AT1-receptors and α1D-adrenoceptor subtypes, which may play an important role in mediating the renal vascular response in this fructose-fed model.
Overall, the results demonstrate that there is a reduction in the sensitivity of the renal vasculature to adrenergic agonists in the hyperinsulinaemic state where there is likely to be a sympatho-excitation. This reduction would appear to result from a decrease in adrenergic receptor sensitivity as well as a reduced responsiveness to Ang II suggestive of a defect in intracellular signalling. Furthermore, despite the fact that the renal α1D-adrenoceptor subtype contributes to vasoconstriction under normal conditions, in the FFR, it seems that other subtypes are involved. The present study revealed that the positive interaction between α1D-adrenoceptor subtype and AT1-receptors in the renal vasculature of normal rats is attenuated upon chronic intake of fructose.
Acknowledgements
M. H. A. is a recipient of Vice Chancellor's Award and USM fellowship from the Institute of Postgraduate Studies (IPS) of Universiti Sains Malaysia, all gratefully acknowledged. The present study was carried out under the support of Research University Grant of Universiti Sains Malaysia to M. A. S. Md. H. A., M. A. S. and E. J. J. contributed to the study design, experiments, interpretation of the results and manuscript preparation. M. A. H. K., N. A. A. and H. A. R. assisted in the biochemical and statistical analysis. There is no conflict of interest.






