


Introduction
Hydrochemical studies of subglacial meltwaters emerging from beneath Arctic glaciers have revealed that significant solute enrichment takes place in rock–water contact environments with little access to atmospheric gases (e.g. Reference Vatne, Etzelmüller, Ødega rd and SollidVatne and others, 1996; Reference Wadham, Hodson, Tranter and DowdeswellWadham and others, 1998; Reference Skidmore and SharpSkidmore and Sharp, 1999). Despite this constraint, chemical weathering rates are often comparable to continental averages (Reference Hodson, Tranter and VatneHodson and others, 2000), which several researchers have suggested reflects the efficacy of microbial catalysis of key rock-weathering reactions beneath the ice (Reference Wadham, Hodgkins, Cooper and TranterWadham and others, 2001; Reference Wynn, Hodson and HeatonWynn and others, 2006, Reference Wynn, Hodson, Heaton and Chenery2007). With increasing isolation, subglacial meltwater chemistries are thought to depend more and more upon the provision of acidity (H+) from the oxidation of sulphides (Reference Raiswell and ThomasRaiswell and Thomas, 1984; Reference Skidmore, Tranter, Tulaczyk and LanoilSkidmore and others, 2010; Wadham and others, in press). Equations (1–3) below show how this oxidation is achieved using a range of oxidants, beginning with the dissolved atmospheric O2 (DO) in the snow- and/or ice melt entering the glacier (Equation (1)). Observations of glacial runoff suggest that SO4 2− concentrations often exceed the maximum value that may be produced by the DO content of meltwaters entering the glacier via crevasses (e.g. Reference Hodson, Tranter and VatneHodson and others, 2000; Reference Tranter, Sharp, Lamb, Brown, Hubbard and WillisTranter and others, 2002), indicating the use of alternative oxidants, as exemplified by Equations (2) and (3).



To date, knowledge concerning the relative importance of these different oxidants remains elusive. Studies from marine environments have shown that these alternatives are kinetically unfavourable unless microorganisms are directly involved (e.g. Reference Schippers and JørgensenSchippers and Jørgensen, 2001). The implication, therefore, is that bacteria are also active under many glaciers with high SO4 2- yields (Reference Sharp, Parkes, Cragg, Fairchild, Lamb and TranterSharp and others, 1999; Reference Skidmore, Anderson, Sharp, Foght and LanoilSkidmore and others, 2005) and that SO4 2- production from suboxic environments is an ideal fingerprint for microbial life between ice sheets (e.g. Reference HodgsonHodgson and others, 2009).
Within the context presented above, it is clear that the subglacial hydrology and biogeochemistry of Arctic glaciers may be better understood if the DO status of meltwaters emerging from glaciers can be linked to the oxygen demands imposed by abiotic and microbially mediated oxidation reactions occurring in different subglacial environments. Here we address this research need by coupling high-resolution time series of total dissolved solids, DO and runoff to sulphide oxidation in a subglacial flowpath beneath Midtre Lovenbreen, Svalbard, during the summer of 2005.
Field Site
Midtre Lovenbreen is a small 5.2 km2 valley glacier in northwest Svalbard (78°54’N, 11°80’E; Fig. 1). Negative mass balance of the glacier has dominated since 1968, with accelerating ice losses reaching the contemporary rates of –0.69 m w.e. a−1 (Reference KohlerKohler and others, 2007). The geology of the catchment can be broadly summarized as metamorphic phyllites and schists, with bands of quartzite, to the south and sandstones, chert and carbonates to the north (Reference HjelleHjelle, 1993). All these rocks contain pyrite and at least trace amounts of carbonate.

Fig. 1. Location map of Midtre Lovénbreen, for 2005, highlighting river gauging stations ( MLE and MLW), upwellings (UPW1 and UPW2), the likely extent of temperate ice at the glacier bed (after Reference BjörnssonBjörnsson and others 1996; Reference RippinRippin, 2002; Reference Zwinger and MooreZwinger and Moore, 2009) and the approximate lower limits of surface micro-catchments that drain into the glacier.
Midtre Lovenbreen is polythermal: temperate ice is sourced in the uppermost cirques (Reference Zwinger and MooreZwinger and Moore, 2009), extending ∼2.5km down-glacier as a layer at the ice-bed interface with a maximum depth of ∼50m (Reference BjörnssonBjornsson and others, 1996; Reference RippinRippin and others, 2003). Annual flow velocities are <8 m a−1 (Reference LiestølLiestøl, 1988; Reference BjörnssonBjornsson and others, 1996; Reference Rippin, Willis and ArnoldRippin and others, 2005). It is where the cirques join the main glacier that surface-derived meltwaters enter the glacier. At lower elevations, meltwaters drain entirely via supraglacial drainage pathways that transfer ∼75% of the annual melt (Fig. 1; Reference Hodson, Kohler and BrinkhausHodson and others, 2005b; Reference Irvine-FynnIrvine-Fynn, 2008). Dye-tracer passage through the en/ subglacial drainage system revealed moderately rapid mean transit velocities of ∼0.3 m s−1, suggesting a typical through-flow time of <5 hours and not unlike the supraglacial streams (Reference Irvine-Fynn, Hodson, Kohler, Porter, Vatne and MavlyudovIrvine-Fynn and others, 2005a). However, annual water budgets for 1997–2002 have indicated runoff yields are also strongly influenced by storage within the glacier’s drainage system, especially before the meltwaters entering the glacier have begun to emerge as a pressurized upwelling in the glacier forefield (Reference Hodson, Kohler and BrinkhausHodson and others, 2005b).
Data Collection
During the ablation season, bulk meltwater from the glacier consistently occupied two key routes: one to the east and one to the west (hereafter MLE and MLW; see Fig. 1). In the summer of 2005, two subglacial upwellings occurred to the east of the glacier centre line (Fig. 1). The first, UPW1, emerged on day of year (DOY) 182 and the second (UPW2) on DOY 213. Druck pressure transducers and Campbell 207 electrical conductivity probes were used to measure hourly averages of hydrological variables in MLE and MLW (see Reference Irvine-FynnIrvine-Fynn and others, 2005b). Uncertainty in discharge, Q, in MLE was <6% and 16% in MLW. Electrical conductivity (manufacturer’s accuracy of ±10%) was also monitored at the artesian discharge point of UPW1. All conductivity series were transformed into continuous total dissolved ionic solutes (TDSi) by regression analysis with an uncertainty of ±1.9%, ±3 . 2% and ±2 . 0% at MLE, MLW and UPW1, respectively. Dissolved O2 was measured alongside TDSi at UPW1 using an OxyGuard CS512 DO sensor (maximum uncertainty <0.2 mgL−1). Sensor drift did not occur, at least according to the periodic DO sensor maintenance undertaken to eliminate biofouling or sediment influence on sensor response.
Sampling of meltwater was undertaken at MLE, MLW, UPW1, and UPW2 at intervals typically <48 hours. Opportunistic sampling was also conducted upon the glacier where meltwaters descended into crevasses. All samples were taken manually and handled according to the protocol detailed by Reference Hodson, Mumford, Kohler and WynnHodson and others (2005a). Samples were filtered within 6 hours of abstraction and then stored, airtight, at 4°C prior to major-ion analysis by Dionex 1CS-90 ion chromatography (precision <10% for solute described here) within 3 months of the field campaign. Concentrations of HCO3 − were estimated using charge balance errors.
Results
Figure 2 includes the time series of hydrometeorological observations made during 2005. Peak Q was reached on day of year (DOY)188, being 2.63 and 1.46 m3 s−1 at MLE and MLW, respectively, coincident with seasonal peak air temperatures. Lower Q, coupled with elevated TDSi, was apparent during a cool period (DOY194-199). Following DOY200 until the termination of observations, MLW was characterized by a stable period of diurnally oscillating discharge with a mean of 0.32 m3s−1. Subglacial waters emerged on DOY182 at UPW1 beginning as a small turbid effusion, but gradually enlarged to an artesian fountain up to ∼0.7m which showed decreasing trend over the latter half of the observation period. The effect of UPW1 was to increase the volume of water passing through MLE compared to that gauged at MLW. The emergence of subglacial water also increased TDSi in MLE by ∼750| μEqL−1 and weakened the correlation between Q and TDSi ( r=–0.4) compared to MLW ( r=–0.6). Figure 2 shows that TDSi at UPW1 displayed far less variability than bulk meltwaters monitored at MLE and MLW. The second upwelling (UPW2) appeared on DOY213 and combined, the upwellings contributed to a mean difference in Q between MLW and MLE of ∼0.6m3s− 1. Interestingly, MLE, MLW and UPW1 all evidenced TDSi peaking at ≥2500 μEqL−1 towards the close of the ablation season. Concentrations of SO4 2− at UPW1 tracked TDSi closely (r=0.89) and contributed up to 980 μEq L−1 (34%) of the ion load at the end of the observation period. There was a remarkable similarity between the SO4 2− content of UPW1 and UPW2 (Fig. 2), which was also the case for the other major ions in subglacial outburst waters (data not shown).

Fig. 2. Time series of: (a) air temperature recorded at 150 m a.s.l. on the glacier snout and precipitation events; (b) Q for MLE (black) and MLW (grey); (c) TDSi at MLE (black), MLW (grey) and UPW1 (dotted); (d) DO recorded at UPW1; and (e) *SO4oxic at UPW1 (line) and observed SO4 2− at UPW1 (dot) and UPW2 (square).
The DO time series is presented in Figure 2 as hourly averages. Dissolved oxygen saturation for waters <1°C, as recorded at UPW1, is 14.1 mgL−1 at typical atmospheric pressure (Reference Lewis and WildeLewis, 2006). Thus, the upwelling waters were initially suboxic (<10mgL−1) and gradually approached supersaturation (∼15mgL−1) on DOY190. Subsequently, as proglacial discharge fell, UPW1 water returned to a suboxic state until ∼DOY205. Following DOY208, the DO time series was characterized by marked diurnal oscillations until DOY230, which were significantly correlated to Q (r= 0.30, p < 0.01), but inversely correlated to TDSi (r=–0.32, p<0.01). A lag time of 3 hours was identified by cross-correlation, with Q leading DO (r=0.39). The initiation of UPW2 on DOY213 appeared to have had no impact upon either the TDSi or DO records at UPW1.
Discussion
The evolution from suboxic initial subglacial outburst waters towards saturation is thought to be an annual occurrence at Midtre Lovenbreen according to spot samples (e.g. Reference Wynn, Hodson and HeatonWynn and others, 2006). However, high-resolution dynamics had yet to be reported at this or any other glacier, although a twice-daily spot sampling campaign has been described at Haut Glacier d’Arolla, Switzerland (Reference Brown, Tranter, Sharp, Davies and TsiourisBrown and others, 1994). There, DO variations were inversely related to Q, both in bulk and supraglacial meltwaters, leading Reference Brown, Tranter, Sharp, Davies and TsiourisBrown and others (1994) to speculate that shorter residence times during peak melt and low ice O2 content control DO concentrations. This is in stark contrast to the present study, since there is a positive correlation between DO and Q and a negative correlation between DO and TDSi (see above). The strong inverse relationship between DO and TDSi in the interval DOY208–230, therefore, suggests that daily variations in the mixing ratio of oxygenated surface meltwaters and suboxic or perhaps even anoxic subglacial waters take place at Midtre Lovenbreen. Seasonal variations in DO also suggest that the mixing ratio of these flow paths changes, perhaps in response to drainage system evolution throughout the summer. Furthermore, the changes contrast markedly to those proposed by Reference Wynn, Hodson and HeatonWynn and others (2006) for this site because suboxic waters are apparent throughout the summer but at a time of the day when Reference Wynn, Hodson and HeatonWynn and others (2006) were not conducting their spot sampling.
Confirmation that surface meltwaters were well oxygenated was acquired by in situ analysis of the supraglacial stream DO levels using Winkler titration. A total of 11 analyses between mid-July and mid-August at a range of sites yielded DO levels of 13.2 ± 1.24 mg L−1 and thus close to saturation. Furthermore, melt experiments using snow, glacier ice and superimposed ice samples in the laboratory indicated supersaturation with DO relative to the atmosphere. The DO levels in five successive melt fractions were 104–120% saturated, the snow samples being responsible for the greatest levels upon initial melting. In addition to the snowmelt source, oxygen supersaturation may occur through turbulent dissolution as meltwaters cascade down the hydrologically active crevasses. Therefore, surface-derived waters at Midtre Lovenbreen almost certainly enter the glacier close to O2 saturation.
Atmospheric sources of SO4 2− cannot explain the high levels observed in Figure 2. For example, SO4 2−/Cl− ratios in UPW1 and UPW2 (not shown) were 50–250 times those typical of the local snowpack according to Reference Hodson, Tranter and VatneHodson and others (2000). The potential for SO4 2− production following Equation (1) was therefore estimated from the depletion of DO in upwelling waters shown in Figure 2, by assuming that surface waters entering the glacier at ∼0°C were saturated with respect to the atmosphere. The resulting SO4 2− quantity, hereafter *SO4 oxic, therefore represents the maximum potential for SO4 2− production by Equation (1) if no other O2 sources or sinks are assumed to exist within the glacier. Figure 2 shows *SO4 oxic alongside the observed SO4 2− at UPW1 and UPW2. Two key observations emerge:
-
1. Aerobic oxygenation of sulphide is an insufficient source of the observed SO4 2− prior to DOY 208 and after DOY 230. During these periods, SO4 2− levels in UPW1 and UPW2 often exceed concentrations achievable with complete consumption of DO in saturated supraglacial meltwater by Equation (1).
-
2. Estimates of *SO4 oxic are close to the observed SO4 2− concentrations during the main phase of the ablation season, when strong diurnal variations in DO were sampled, usually at the time of high DO in the afternoon.
Given the above, it is tempting to propose that aerobic oxidation of sulphide minerals is a sufficient explanation for the mid-season levels of SO4 2− observed in subglacial outflow, at least during peak flows. An analysis of the kinetics of SO4 2− acquisition over timescales relevant to meltwater passage through the glacier was therefore undertaken. Table 1 shows the chemistries of solutions produced in three successive, 24 hour, closed system dissolution experiments at high rock–water contact ratios (1g L−1) of <125 μm freshly crushed bedrock and deionized water. These samples were analysed in the manner described above. All experiments clearly show the effects of carbonate weathering, producing solutions with high Ca2+, low p(CO2) and saturation with respect to CaCO3 (Table 1). This is typical of early rock-water contact beneath glaciers (e.g. Reference Tranter, Sharp, Lamb, Brown, Hubbard and WillisTranter and others, 2002). However, there is a notable lack of SO4 2− in the solutions, such that <1% of the carbonate weathering can be attributed to the neutralization of acidity generated by sulphide oxidation (using Equation (1)). By contrast, the corresponding proportions in the UPW1 and UPW2 samples were approximately 48±7.3% and 52 ±8.9%, respectively (Reference Irvine-FynnIrvine-Fynn, 2008).
Table 1. Major-ion, pH, CO2 partial pressure (p(CO2)), sulphate mass fraction (SMF) and saturation index (SI) calculations (produced using Phreeqc software) for CaCO3 following the dissolution experiments. Experimental conditions are described in the text and more closely by Reference HodsonHodson and others (2010). Three successive 24 hour experiments are reported for each major rock type: siliceous sandstone (SSt), phyllite (Phyll), schist/phyllite (Sch/Phyll) and quartzite (Qtz)

Following the above argument, aerobic oxidation of sulphide mineral surfaces from suspended sediments as surface waters transit through the en/subglacial drainage system (∼5 hours according to dye injections) cannot explain the SO4 2 - observed at UPW1 and UPW2. This necessitates an explanation based upon mixing between surface-derived meltwaters and long-residence-time waters draining reactive subglacial sediments throughout the summer. The kinetics of sulphide oxidation during our dissolution experiments suggest that abiotic oxidation remains unlikely. Furthermore, stable-isotopic work at this site has already identified that microbially mediated sulphide oxidation occurs in anoxic environments within the local glacial sediments (Reference Wynn, Hodson and HeatonWynn and others, 2006). However, since other bacterial processes are also known to occur where these sediments are not anoxic (e.g. nitrification (Reference Hodson, Mumford, Kohler and WynnHodson and others, 2005a; Reference Wynn, Hodson, Heaton and CheneryWynn and others, 2007)), it seems reasonable to expect aerobic sulphide oxidation is also being mediated by bacteria.
During the present study, far greater NO3 −/Cl− molar ratios were observed in UPW1 and UPW2 waters (averaging 0.054 ± 0.011 for all samples) relative to supraglacial samples of waters descending crevasses (average NO3 −/Cl− = 0.009 ±0.018). Microbial NO3 − production therefore appears to have been significant. Furthermore, a positive correlation (r=0.85) between the NO3 −/Cl− ratios of the UPW1 and UPW2 waters and their SO4 2− concentrations seems to rule out denitrification (Equation (3)) as a basis for providing the additional SO4 2− not explained by Equation (1). Consequently, we tentatively propose that transition metals such as Fe(III) (Equation (2)) and Mn(IV) (Equation (4); Reference Schippers and JørgensenSchippers and Jørgensen, 2001) are the most likely oxidants responsible for sulphide oxidation in the anoxic environments:
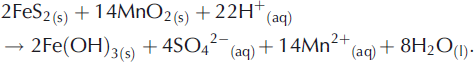
However, since the isotopic analysis of Reference Wynn, Hodson and HeatonWynn and others (2006) does not indicate the anoxic production of SO4 2− during the principal runoff phase (e.g. DOY 208–230), further research into the timing and relative importance of aerobic versus anoxic SO4 2− production appears well justified.
Conclusion
For the first time, a high-resolution DO record has been acquired from subglacial water emerging from Midtre Lovénbreen, an Arctic glacier. This record shows a clear distinction between the polythermal glacier and a temperate glacier, because surface meltwaters are highly oxygenated until they reach the glacier bed, where a clear oxygen demand is present owing to sulphide oxidation. While the same oxygen demand is known beneath temperate glaciers, the impact on DO is apparently masked by low DO levels in ice melt upon the surface and perhaps their re-aeration in turbulent transit through the glacier. The Arctic glacier differs from the temperate glacier on account of a subglacial, rather than supraglacial, control upon DO dynamics in runoff from beneath the glacier. The present study shows the subglacial acquisition of SO4 2− by sulphide oxidation has the capacity to account for more than all of the oxygen depletion observed during the summer. It is, therefore, necessary to invoke microbial catalysis of sulphide oxidation, perhaps in both aerobic and anoxic environments, because the kinetics of abiotic SO4 2− production in dissolution experiments are very slow. Where oxidants other than atmospheric O2 are active, the use of transition metals appears more likely than NO3 − because high SO4 2− concentrations in subglacial waters are concomitant with high NO3 − following nitrification.
Acknowledgements
T.I.-F. acknowledges UK Natural Environment Research Council (NERC) standard grant NE/G006253/1 and financing of fieldwork from the Gino Watkins Memorial Fund, the Dudley Stamp Memorial Trust and the University of Sheffield (Department of Geography). A.J.H. acknowledges the University of Sheffield (Knowledge Transfer Fund), the Geological Society’s W.G. Fearnsides Fund and the Earth and Space Foundation. N. Cox and S. Marshall (NERC Arctic Research Station), P. Porter and F. Hunter helped with the fieldwork. Dairy Pipe Lines Ltd (Saffron Walden, Essex) are thanked for donation of beverage-grade stainless steel mesh used to house the DO probe. M. Tranter provided useful review comments.



