


Hypoxia is a common characteristic of all solid tumours (Ref. Reference Vaupel, Hockel and Mayer1) and is a condition in which proliferating tumour cells are deprived of oxygen due to a limited blood supply from abnormal tumour microvasculature (Fig. 1). Hypoxic cells are at risk of stress-induced insults including oxidative DNA damage, DNA strand breaks and genetic aberrations, which can restrain growth and ultimately result in cell death. However, cancer cells show a range of genetic changes that improve survival and enable them to adapt to hypoxic conditions. As a result, hypoxic tumour cells continue to proliferate, are associated with a more invasive and metastatic phenotype and are usually resistant to conventional treatments such as radiotherapy and chemotherapy (Refs Reference Hockel2, Reference Hockel3, Reference Hockel4, Reference Hockel and Vaupel5, Reference Hockel6). Therefore, gaining a clearer understanding of the underlying molecular mechanisms involved in hypoxia signalling in cancer cells and how these processes might become deregulated in different cancer types is likely to result in better targeting of hypoxic tumour cells and more-effective treatments for solid tumours. A central component of hypoxia signalling in the cell is hypoxia-inducible factor (HIF), which is critically involved in both sensing and responding to changes in cellular oxygen (Ref. Reference Wang7). This review outlines the therapeutic strategies used to target HIF/hypoxic signalling in tumour cells and discusses recent advances made in this area.

Figure 1. Increased hypoxia is detected at increasing tumour size. The figure shows fluorescence imaging of human glioblastoma xenograft tumours. Tumours from the human glioblastoma multiforme tumour cell line E106 were transplanted subcutaneously into nude mice. Tumours were harvested when they reached a mean size of 2 mm (a), 4 mm (b), 6 mm (c), 8 mm (d) and 10 mm (e). Tumour sections were stained with antipimonidazole (green) to indicate areas of hypoxia, 9F1 (a monoclonal antibody to mouse endothelium; red) to assess vasculature, and Hoechst 33324 (blue) for nuclei. As tumour size increased, the vascular architecture became less organised. This figure was kindly provided by Dr Jan Bussink (Department of Radiation Oncology 874, Radboud University Nijmegen, The Netherlands) and is reprinted from Ref. Reference Hendriksen153, with permission from Elsevier (© 2009 Elsevier).
The HIF pathway
HIF is a transcriptional complex activated in response to changes in cellular oxygen levels and mediates the expression of many genes (Refs Reference Guillemin and Krasnow8, Reference Semenza9). HIF target genes encode proteins that are involved in the regulation of various aspects of tumour biology, including oxygen transport, iron metabolism, glycolysis, glucose transport, cell survival and proliferation, angiogenesis, invasion and metastasis. HIF activity is deregulated in many human cancers, and this is most commonly due to the overexpression of HIF-α, the regulatory subunit of the HIF complex. Overexpression of HIF-α is usually associated with increased vascular density, severity of tumour grade, treatment failure and a poor prognostic outcome (Refs Reference Bos10, Reference Zhong11). Blocking HIF activity or targeting HIF-1α expression in tumours has been shown to significantly slow tumour growth in xenograft models (Ref. Reference Maxwell12) and render hypoxic cells more susceptible to killing by conventional therapies (Refs Reference Staab13, Reference Williams14, Reference Moeller15, Reference Moeller16).
HIF-1: structure and regulation
The HIFs belong to a family of structurally related basic-helix–loop–helix (bHLH)-containing proteins (Ref. Reference Wang7). The prototype of the family is HIF-1. HIF-1 consists of two subunits: the regulatory HIF-1α subunit and the ubiquitously expressed HIF-1β subunit (also known as aryl hydrocarbon receptor nuclear translocator, ARNT). HIF-1α protein is composed of four functional domains: a bHLH domain and a PER–ARNT–SIM (PAS) domain (involved in dimerisation and DNA binding), an oxygen-dependent degradation (ODD) domain (required for targeting to the proteasome and degradation), and two transactivation domains (N-TAD and C-TAD) required for transcriptional activation (Ref. Reference Jiang17) (Fig. 2). HIF-1β contains bHLH, PAS and transactivation domains (Ref. Reference Li, Ko and Whitlock18).

Figure 2. Schematic representation of HIF-1α and its four functional domains. The basic-helix–loop–helix (bHLH) and PER–ARNT–SIM (PAS) domains of hypoxia-inducible factor 1α (HIF-1α) are involved in dimerisation and DNA binding; the oxygen-dependent degradation (ODD) domain is required for degradation via the proteasome; and the transactivation domains (N-TAD and C-TAD) are involved in transactivation activity. Hydroxylation of proline residues 402 and 564 within the ODD domain mediates its interaction with the von Hippel–Lindau tumour suppressor protein (pVHL). Hydroxylation of asparagine 803 in the C-TAD blocks its association with transcriptional coactivator p300/CBP.
Whereas HIF-1β is constitutively expressed in cells, the availability of HIF-1α is dependent on cellular oxygen levels. In normoxia (21% oxygen levels), HIF-1α protein is rapidly and continuously expressed and degraded (for review, see Ref. Reference Semenza9). The synthesis of HIF-1α protein is regulated by oxygen-independent mechanisms involving growth-factor-mediated activation of the phosphoinositide 3-kinase (PI3K) and mitogen-activated protein kinase (MAPK) pathways (Refs Reference Fukuda19, Reference Laughner20). HIF-1α protein degradation is controlled by the ODD domain, and deletion of the entire ODD region renders HIF-1α stable even in the absence of hypoxia signalling (Ref. Reference Huang21). Hydroxylation of proline residues 402 and 564 within the ODD domain of HIF-1α mediates its interaction with the von Hippel–Lindau tumour suppressor protein (pVHL), which is the recognition component of an E3 ubiquitin ligase, leading to HIF-1α ubiquitination and subsequent degradation by the 26S proteasome (Refs Reference Ivan22, Reference Jaakkola23, Reference Masson24, Reference Yu25). The hydroxylation process is governed by three evolutionarily conserved HIF prolyl hydroxylases – PHD1 (EGLN2), PHD2 (EGLN1) and PHD3 (EGLN3) – and their activity depends on the availability of oxygen, iron, 2-oxoglutarate and ascorbate (Refs Reference Bruick and McKnight26, Reference Epstein27). Interestingly, it has been shown by using small interfering RNA (siRNA) techniques that PHD2 plays a predominant role in controlling HIF-1α levels (Ref. Reference Berra28).
Under hypoxic conditions, prolyl hydroxylation within the ODD domain is inhibited and the interaction of HIF-1α with pVHL is prevented. As a result, HIF-1α ubiquitination and degradation is blocked and consequently the level of the protein increases. The accumulated HIF-1α translocates to the nucleus where it dimerises with HIF-1β (Ref. Reference Wang7) via the bHLH and part of the PAS domain to form the HIF-1 complex. HIF-1 recruits transcriptional coactivators such as p300/CBP (p300/CREB-binding protein) (Ref. Reference Kallio29) and binds to the hypoxia-response element (HRE) within the promoter region of HIF-1-responsive target genes (Ref. Reference Wang and Semenza30), thereby mediating their transcriptional activation.
The transcriptional activity of HIF-1 is also controlled by an asparagine hydroxylase known as FIH-1 (factor inhibiting HIF-1) (Refs Reference Hewitson31, Reference Lando32, Reference Lando33, Reference McNeill34, Reference Mahon, Hirota and Semenza35). In normoxia, hydroxylation of Asn803 in the C-TAD of HIF-1α blocks its association with p300/CBP. FIH-1 was also reported to interact with pVHL to modulate HIF-1α protein stabilisation (Ref. Reference Mahon, Hirota and Semenza35). Thus, FIH-1 acts as a negative regulator of HIF-1α to suppress transcriptional activity.
Other HIF-α family members
Two other HIF-α isoforms have been identified: HIF-2α and HIF-3α (Ref. Reference Semenza36). HIF-2α has a similar structure to HIF-1α (Ref. Reference Bardos and Ashcroft37). Like HIF-1α, HIF-2α is rapidly induced in response to hypoxia, negatively regulated by the pVHL ubiquitin E3 ligase complex, and can mediate the transcriptional activation of a number of known HIF-1 target genes (Ref. Reference Wiesener38). However, the expression of HIF-2α is cell-type specific and it has a distinct biological role from HIF-1α (Refs Reference Carroll and Ashcroft39, Reference Compernolle40, Reference Hu41, Reference Krieg42, Reference Raval43), with particular importance in renal cancer and vascular biology. In development, HIF-1α and HIF-2α were demonstrated to have nonoverlapping functions: HIF-1α−/− and HIF-2α−/− mouse embryos have different phenotypes and developmental defects (Ref. Reference Compernolle40). HIF-2α is also expressed at a higher level than HIF-1α in several pVHL-defective renal carcinoma cell lines (Refs Reference Krieg42, Reference Maxwell44), and various groups have reported that HIF-1 and HIF-2 can regulate both overlapping and distinct target genes (Refs Reference Carroll and Ashcroft39, Reference Hu41, Reference Raval43).
The function of HIF-3α is not well understood. Several splice variants of HIF-3α have been identified (Ref. Reference Maynard45). One of the HIF-3α splice variants, known as the inhibitory PAS domain protein (IPAS), can function as a dominant negative regulator of hypoxia-inducible gene expression: it binds to the HIF-1α subunit to form a nonfunctional complex in the nucleus, impairing the expression of HIF-1 target genes under hypoxic conditions (Ref. Reference Makino46). Interestingly, it has been reported recently that IPAS gene expression is induced in response to hypoxia and is regulated directly by HIF-1 binding, forming a further level of negative feedback in the hypoxia-response system (Ref. Reference Makino47).
HIF and cancer
In addition to hypoxia, loss-of-function mutations in several genes involved in the oxygen-sensing mechanism have also been shown to contribute to the overexpression of HIF-α (HIF-1α and HIF-2α) and activation of the HIF pathway in tumour cells (Fig. 3). For example, loss-of-function mutations in VHL have been shown to increase HIF-1α and HIF-2α expression in clear-cell renal carcinoma, haemangioblastoma and other VHL-associated tumours due to the lack of HIF-α ubiquitination and degradation (Ref. Reference Maxwell44). Mutations in succinate dehydrogenase (SDH) and fumarate hydratase (FH) inhibit prolyl hydroxylase activity, resulting in abnormal stabilisation of HIF-1α and upregulation of HIF target genes such as vascular endothelial growth factor (VEGF) in several cancers, namely paragangliomas, phaeochromocytomas, leiomyomas and renal cell cancers (Refs Reference Briere48, Reference Pollard49).
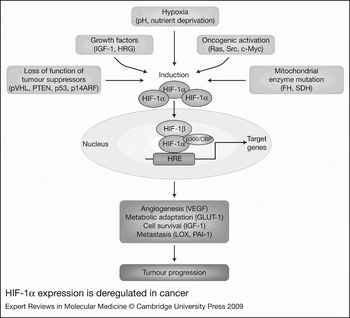
Figure 3. HIF-1α expression is deregulated in cancer. Overexpression of hypoxia-inducible factor 1α (HIF-1α) and activation of the HIF pathway in cancer is caused by a combination of microenvironmental changes, such as changes in oxygen levels (hypoxia), pH and nutrients (deprivation), increases in growth factors, and genetic abnormalities leading to loss of tumour suppressor function, oncogenic activation or deregulated mitochondrial function. Increased HIF-α protein in cancer cells translocates to the nucleus, binds to HIF-1β, recruits coactivators (e.g. p300/CBP) and activates the transcription of multiple genes involved in angiogenesis (e.g. VEGF), metabolic adaptation (e.g. GLUT-1), cell survival (e.g. IGF-1) and metastasis (e.g. LOX, PAI-1) – thereby driving tumour progression. Abbreviations: FH, fumarate hydratase; GLUT-1, glucose transporter 1; HRG, heregulin; IGF-1, insulin-like growth factor 1; LOX, lysyl oxidase; p300/CBP, p300/CREB-binding protein; p14ARF, alternate reading frame (ARF) product of CDKN2A (cyclin-dependent kinase inhibitor 2A) locus; PAI-1, plasminogen activator inhibitor 1; PTEN, phosphatase and tensin homologue; pVHL, von Hippel–Lindau tumour suppressor protein; SDH, succinate dehydrogenase; VEGF, vascular endothelial growth factor.
Dysregulation of key signal transduction pathways also contributes to the overexpression of HIF-1α and activation of HIF-1 in cancer. Tumour cells with constitutive activation of the Ras–MAPK pathway (Ref. Reference Berra, Pages and Pouyssegur50), Src (Ref. Reference Karni51) or the PI3K–AKT(PKB)–mTOR (mammalian target of rapamycin) pathway (Refs Reference Lee52, Reference Zhong53) have elevated expression of HIF-1α protein. Loss of function of tumour suppressor proteins such as PTEN (which leads to constitutive activation of AKT) (Refs Reference Bardos and Ashcroft54, Reference Zundel55) and p53 can also result in increased HIF-1 activity (Fig. 3).
Since HIF-α (HIF-1α and HIF-2α) is induced in cancer cells in response to hypoxia and growth factors, and as a result of known genetic abnormalties, it is no surprise then that HIF-α protein has been shown to be overexpressed in human tumour biopsy samples. Immunohistochemical analyses of paraffin-embedded tissue sections have shown HIF-1α (nuclear) to be highly expressed in many tumour types including pancreatic (Ref. Reference Miyake56), head and neck (Refs Reference Koukourakis57, Reference Winter58), oropharyngeal (Ref. Reference Aebersold59), breast (Refs Reference Bos60, Reference Gruber61), renal (Ref. Reference Klatte62), ovarian (Ref. Reference Osada63), urothelial (Ref. Reference Ke64), bladder, brain, colorectal and prostate (Ref. Reference Talks65). Several independent studies have revealed a strong correlation between HIF-1α overexpression and patient mortality. High HIF-1α expression has also been associated with low survival rates in pancreatic carcinoma (Ref. Reference Miyake56), head and neck squamous cell carcinoma (Ref. Reference Winter58), clear-cell renal cell carcinoma (Ref. Reference Klatte62) and breast carcinoma (Refs Reference Bos60, Reference Gruber61). This might be because the overexpression of HIF-1α, which often indicates significant levels of tumour hypoxia, is involved in mediating cellular adaptive responses that enable tumour cells to survive. Tumour hypoxia and HIF-1α overexpression is reported to correlate with an increased aggressiveness of tumour cell behaviour, angiogenesis (Ref. Reference Koukourakis57) and metastasis (Ref. Reference Gruber61) and can be used as a marker to predict outcome in patients with metastatic disease. Interestingly, a study in clear-cell renal cell carcinoma has shown that HIF-1α expression directly correlates with markers of apoptosis (p53) and growth inhibition (p21), the mTOR pathway (AKT, p27), the chemokine receptors CXCR3 and CXCR4, and proteins of the VEGF family (Ref. Reference Klatte62). Therefore, induction of HIF-1α in many cancer types results in several consequences that could enable tumour cells to survive and continue to proliferate.
Surprisingly, not all tumours that exhibit HIF-1α overexpression have been found to be associated with decreased patient survival rates (Ref. Reference Fillies66). For example, in early-stage squamous cell carcinoma of the oral floor HIF-1α overexpression is associated with improved survival rates (Ref. Reference Fillies66). This difference may arise from the fact that HIF-1α could function by having a dual role in early carcinogenesis. On the one hand, HIF-1α promotes tumour angiogenesis and cell survival when mediating an adaptive response, while on the other hand, in response to cellular stress HIF-1α cooperates with the apoptotic machinery (via induction of apoptotic genes or crosstalk to p53) to mediate tumour cell death (Ref. Reference Sumiyoshi67). Indeed, the function of the HIFs in tumour progression might depend on the cell type and cellular context as well as the stage of carcinogenesis, and further work is needed to clarify this in order to establish when best to target the HIF pathway in cancer and whether certain cancer types would prove more or less sensitive to a HIF inhibitor.
Strategies to target the HIF pathway in cancer
In recent years, several strategies have been developed to identify direct and indirect inhibitors of HIF-α that function by blocking HIF-α (HIF-1α or HIF-2α) expression levels and/or HIF (HIF-1 or HIF-2) activity. These include cell-based reporter screens, antisense approaches, targeting key protein–protein interactions, increasing HIF-1α protein turnover or utilising a HIF oligonucleotide decoy (Table 1). In addition, therapeutic exploitation of other key pathways and mechanisms that are known to regulate HIF-1α protein availability (stability and synthesis) and HIF-1 activity could also potentially be utilised to target the HIF pathway in cancer (Fig. 4). While the HIF transcriptional complex itself is a challenging therapeutic target, blockade of the HIF pathway and inhibition of HIF-α expression is therapeutically attractive because of its pivotal role in driving angiogenesis and tumour progression. Overexpression of HIF-1α in many cancers and deregulation of HIF activity offers a degree of selectivity for tumour cells over normal tissue (Ref. Reference Maxwell68), and blocking HIF-1α especially when in combination with conventional therapies has a significant impact on tumour growth (Ref. Reference Bastien69).

Figure 4. Strategies to target the HIF-1 pathway in cancer. (a–c) Several strategies to specifically target hypoxia-inducible factor 1α (HIF-1α) protein levels and HIF-1 activity in cancer cells have been developed (shown in red), including: (a) inhibition of protein–protein interactions (e.g. HIF-1 dimerisation or coactivator recruitment); (b) inhibition of HRE transcriptional activity (e.g. using small-molecules identified in HRE cell-based reporter screens); and (c) activation of HIF-1α protein degradation (e.g. using PHD activators). (d–f) In addition, therapeutic targets currently in development that are known regulators of the HIF-1 pathway provide an alternative means for blocking HIF-1α protein availability (stability and synthesis) and HIF-1 activity in tumour cells (shown in green), including: (d) inhibition of signalling pathways upstream of HIF (PI3K–AKT–mTOR/HDM2 and Ras–MAPK); (e) inhibition of chaperone proteins (e.g. HSP90); and (f) activation of the tumour suppressor p53. Direct downstream targets are indicated by solid lines, and downstream effectors are indicated by dashed lines. Abbreviations: AKT, AKT/protein kinase B; HDM2, human homologue of MDM2 (E3 ubiquitin ligase; p53-binding protein); HRE, hypoxia-response element; HSP90, heat shock protein 90; MAPK, mitogen-activated protein kinase (also known as extracellular-signal-regulated kinase, ERK); MEK, MAPK kinase; mTOR, mammalian target of rapamycin; p300/CBP, p300/CREB-binding protein; PHD, prolyl hydroxylase domain protein; PI3K, phosphoinositide 3-kinase; pVHL, von Hippel–Lindau tumour suppressor protein; RTK, receptor tyrosine kinase; SIAH1a/2, seven in absentia homologue 1a/2; Ub, ubiquitination.
Table 1. Strategies to identify inhibitors of HIF-1α and the HIF pathway
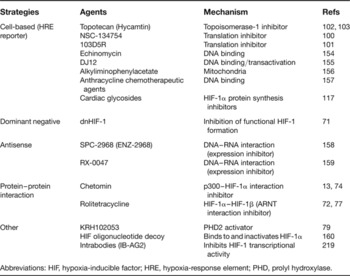
Abbreviations: HIF, hypoxia-inducible factor; HRE, hypoxia-response element; PHD, prolyl hydroxylase.
Targeting HIF-1α directly
As it functions as part of a transcriptional complex, targeting HIF-1α directly is challenging. Specific antisense approaches have been used to reduce HIF-1α expression and transcriptional activity (Ref. Reference Yeo, Chun and Park70), and a dominant negative form of HIF-1α has also been used (Ref. Reference Chen71). Another approach is to inhibit HIF-1 transcriptional activity by blocking HIF-1α protein–protein interactions (Ref. Reference Zinzalla and Thurston72). For example, the binding between HIF-1α and the coactivator p300/CBP, and hence hypoxia-inducible transcription, has been attenuated by retroviral expression of a polypeptide (Ref. Reference Kung73), by the small-molecule chetomin (Refs Reference Staab13, Reference Kung74) or by the use of the indazole compound YC-1 (Refs Reference Li75, Reference Yeo76). In addition, small molecules such as rolitetracycline (a semisynthetic pyrrolidnomethyltetracycline) that block HIF-1α–HIF-1β dimerisation by targeting the PAS domain offer a strategy to block HIF-1-mediated activity in tumour cells by inhibiting the formation of the HIF-1 complex (Ref. Reference Park77).
Targeting HIF-1α expression and/or HIF-1 activity indirectly
Prolyl hydroxylases
Mechanisms that regulate HIF-1α protein stability provide indirect means to target HIF-1α protein levels in tumour cells (Fig. 4). For example, overexpression of PHDs (Ref. Reference Erez78) enhances HIF-1α protein turnover and results in reduced HIF-1α protein availability in tumour cells. Thus, small-molecule activators of the PHDs – such as KRH1020053 – have been developed to reduce HIF-1α protein levels in tumour cells (Ref. Reference Choi79). Alternatively, genetic blockade of SIAH2, which encodes an E3 ubiquitin ligase that ubiquinates PHD2 in hypoxia, leads to reduced HIF-1α availability (Ref. Reference Nakayama80) and provides another potential mechanism for targeting HIF-1α protein stability (Ref. Reference Moller81).
The p53 tumour suppressor protein
Understanding the relationship between HIF and other key signalling pathways can provide valuable therapeutic insight for developing strategies to target HIF indirectly (Fig. 4). For example, considerable progress has been made to our understanding of the molecular mechanisms by which HIF is regulated by the tumour suppressor protein p53, a transcription factor that plays a crucial role in monitoring cellular integrity. When the cell is stressed, p53 protein rapidly accumulates leading to either cell cycle arrest or apoptosis. However, p53 is mutated in over 50% of human cancers (Ref. Reference Soussi and Lozano82). Mutated p53 is unable to transactivate downstream targets and is associated with malignant progression and metastasis (Refs Reference Levine83, Reference Vousden and Lane84). While hypoxia induces cells to undergo p53-dependent apoptosis under some circumstances, cancer cells with dysregulated p53 function are able to survive (Refs Reference Denko85, Reference Giaccia and Kastan86, Reference Graeber87).
p53 is involved in negatively regulating HIF-1α expression and HIF-1 activity (Refs Reference An88, Reference Blagosklonny89, Reference Ravi90, Reference Yang91). While the molecular crosstalk between HIF-1 and p53 is complex, HIF-1α has been observed to bind to p53 in some cellular settings (Ref. Reference An88). An in vitro study has provided biophysical evidence supporting the direct binding of p53 with HIF-1α via the ODD domain within HIF-1α (Ref. Reference Sanchez-Puig, Veprintsev and Fersht92). This interaction was originally proposed to result in p53 stabilisation (Ref. Reference An88) as well as inhibition of HIF-1 activity (Refs Reference An88, Reference Blagosklonny89, Reference Sanchez-Puig, Veprintsev and Fersht92, Reference Schmid93). However, further work has shown that HIF-1α induced in hypoxia does not affect p53 stabilisation (Ref. Reference Ravi90), and it has been proposed that the direct interaction of p53 with HIF-1α leads to HDM2-mediated degradation of HIF-1α (Ref. Reference Ravi90), resulting in hypoxia-induced p53-dependent apoptosis (Refs Reference An88, Reference Blagosklonny89, Reference Chen94). Indeed, recent work has shown that the apoptotic function of p53 can be regulated by the status of HIF-1α in cells and that blocking HIF-1α expression can drive p53-mediated tumour cell death in hypoxia (Ref. Reference Yang91).
It has been suggested that p53 can block HIF-1 transcriptional activity by competing with HIF-1α for p300 (Ref. Reference Schmid93). p53, which itself is a transcription factor, requires the recruitment of p300 for its activity. Using an HRE–luciferase reporter assay to measure the transactivation of HIF-1, it has been demonstrated that low levels of exogenous p53 can block HIF-1 transcriptional activity and this effect can be relieved by overexpression of p300 in the cell (Ref. Reference Schmid93). This competitive binding of p53 for p300 was also confirmed in a separate in vitro transcription assay (Ref. Reference Schmid93). While low levels of p53 can affect HIF-1 activity, high levels of p53 have been reported by several independent studies to block HIF-1α protein accumulation (Refs Reference Bardos and Ashcroft54, Reference Ravi90, Reference Schmid93, Reference Chen94). Furthermore, loss of p53 has been observed to correlate with increased HIF-1α protein level and increased HIF-1 activity (Ref. Reference Ravi90). Forced expression of HIF-1α in p53-expressing tumour cells upregulates VEGF expression (Refs Reference Ravi90, Reference Nieminen95), neovascularisation and the growth of tumour xenografts (Ref. Reference Ravi90). Taken together, these studies clearly demonstrate that p53 negatively regulates HIF-1 transcriptional activity and HIF-1α protein levels and highlights the possibility that reactivating p53 may provide a means to target the HIF pathway in cancer (Fig. 4).
Efforts have been made to identify agents that reactivate mutant p53 (Refs Reference Bykov and Wiman96, Reference Foster97) or activate wild-type p53 (Refs Reference Issaeva98, Reference Vassilev99) in cancer cells (Table 2). Recently, a small-molecule activator of p53, RITA (reactivation of p53 and induction of tumour cell apoptosis), was demonstrated to induce and activate p53, resulting in tumour cell apoptosis (Refs Reference Yang91, Reference Issaeva98). Interestingly, RITA was also observed to block HIF-1α expression, resulting in downregulation of VEGF expression, and antiangiogenic effects in vivo (Ref. Reference Yang91). The ability of a single agent to activate p53-dependent apoptosis and simultaneously suppress tumour angiogenesis represents a novel and promising therapeutic strategy for targeting the HIF pathway in solid tumours (Ref. Reference Yang91).
Table 2. Anticancer agents that decrease HIF-1α and target the HIF-1 pathway

Abbreviations: CDK, cyclin-dependent kinase; COX-2, cyclooxygenase 2; EGFR, epidermal growth factor receptor; HIF, hypoxia-inducible factor; HSP90, heat shock protein 90; MAPK, mitogen-activated protein kinase; 2-ME2, 2-methoxy estradiol; mTOR, mammalian target of rapamycin; PI3K, phosphoinositide 3-kinase; RITA, reactivation of p53 and induction of tumour cell apoptosis; VEGFR, vascular endothelial growth factor receptor.
Other mechanisms for targeting HIF
Several high-throughput cell-based screening approaches using HRE–luciferase reporter systems have identified numerous HIF-1α small-molecule inhibitors that block tumour cell growth by blocking HIF-1α protein accumulation and HIF activity (Refs Reference Chau100, Reference Tan101), although their mechanism of actions remain to be understood. For example, topotecan – a topoisomerase inhibitor and known cytotoxic agent (Table 2) – was found to block HRE activity in a cell-based screen and was subsequently shown to suppress HIF-1α protein translation (Refs Reference Rapisarda102, Reference Rapisarda103). This inhibitor is currently being launched in the clinic to target ovarian and small-cell lung cancer cells. Interestingly, PX-478, another inhibitor of HIF-1α protein translation (although not identified through a cell-based HRE–luciferase reporter screen) is in Phase I clinical trials (Refs Reference Koh104, Reference Macpherson and Figg105, Reference Welsh106); however, the precise cellular target of PX-478 that is responsible for HIF-1α inhibition is not certain, and thus the development of clear clinical endpoints may prove challenging.
In addition to strategic efforts being developed to target the HIF pathway, many recognised anticancer drugs that target known regulators of HIF function have also been shown to block HIF-1α protein levels and/or HIF-1 activity (Table 2). For example, the histone deacetylase (HDAC) inhibitors trichostatin A and FK228 inhibit HIF-1α induction and HIF-1 activity (Refs Reference Manabe107, Reference Mie Lee108, Reference Sasakawa109, Reference Sasakawa110, Reference Yang111). Geldanamycin and 17-AAG, which are HSP90 antagonists, are also effective at inhibiting HIF-1α expression levels (Refs Reference Alqawi, Moghaddas and Singh112, Reference Isaacs113, Reference Kim114, Reference Lang115, Reference Liu116), and recent work has identified digoxin as a potent inhibitor of HIF-1α synthesis and tumour growth (Ref. Reference Zhang117). Other tractable HIF regulators that have emerged recently include c-Myc (Refs Reference Gordan118, Reference Gordan119), c-Met (Refs Reference Chen120, Reference Hara121, Reference Hayashi122) and components of the Ras (Ref. Reference Lim123) and Wnt (Refs Reference Giles124, Reference Jiang125, Reference Kaidi, Williams and Paraskeva126) pathways. A thorough evaluation of how these regulators influence HIF function in cancer may provide further insights into targeting the HIF pathway.
Which HIF-α subunit to inhibit?
When developing new agents to target HIF-α in cancer, the specific effects mediated by a particular HIF-α (HIF-1α or HIF-2α) isoform in different cell types should be taken into consideration (Ref. Reference Carroll and Ashcroft39). Targeting a single HIF-α subunit may not necessarily give the desired effects because it has been demonstrated that different subunits may play distinct roles in different cellular contexts (Refs Reference Carroll and Ashcroft39, Reference Hu41, Reference Covello, Simon and Keith127, Reference Hu128). For example, using siRNA techniques, HIF-1α was reported to be the primary hypoxia-induced transcription factor in breast carcinoma and endothelial cells (Ref. Reference Sowter129), whereas in renal carcinoma cells HIF-2α was shown to be primarily responsible for the induction of hypoxic genes (Ref. Reference Carroll and Ashcroft39). These findings were supported by two separate studies that confirmed HIF-1α primarily regulates the transcription of hypoxia-regulated target genes in MCF-7 breast carcinoma cells, while HIF-2α controls the transcription of target genes such as glucose transporter 1 (GLUT-1) as well as tumour progression in renal carcinoma cells that have lost pVHL function (Refs Reference Carroll and Ashcroft39, Reference Raval43, Reference Rankin130). Interestingly, in pVHL-defective renal carcinoma cells, HIF-1α was found to play a tumour suppressor role. Because the tumour-promoting HIF-α subunit in these cell lines is HIF-2α (Refs Reference Raval43, Reference Gordan118, Reference Rankin130), treatment targeting this subunit may be more beneficial (Ref. Reference Carroll and Ashcroft39). It is important to assess whether targeting both HIF-1α and HIF-2α or either subunit selectively will provide better therapeutic effects in vivo.
Recent work has revealed that pVHL-defective renal carcinoma cells can be further subdivided into tumours with detectable HIF-1α and HIF-2α, or just HIF-2α exclusively (Ref. Reference Gordan119). Accordingly, tumours with detectable HIF-1α and HIF-2α exhibit enhanced activation of AKT–mTOR and MAPK pathways and γH2AX (phosphorylated histone H2AX) accumulation, whereas tumours with only HIF-2α expression display increased c-Myc activity. The identified oncogenic pathways associated with these tumours may enable the strategic selection of combined targeted therapies to be used against these different tumour subtypes.
Translation of HIF inhibitors into the clinic
Hypoxic tumours are usually resistant to radiotherapy, as a result of the low level of oxygen molecules available to generate DNA strand breaks, and to chemotherapy, because of their slow divisional rate, abnormal vasculature and the upregulation of many genes that contribute to their aggressive phenotype. However, blocking HIF-1α renders tumour cells more susceptible to radiotherapy and conventional chemotherapeutic agents (Refs Reference Staab13, Reference Williams14). The recent advances to our understanding of the HIF pathway have helped us to clarify its role in cancer and consequently enable the identification and design of novel therapies. Inhibitors that target the HIF pathway directly or indirectly are attractive and should prove useful in the treatment of most solid tumours including breast, prostate and renal carcinomas. Understanding the mechanism of action of different HIF pathway inhibitors is of particular importance when deciding when and how they would best be used in combination.
Which cancer types to target?
HIF-1α protein levels can be used as a prognosis marker in various cancers (Refs Reference Silva131, Reference Tzao132, Reference Uehara133, Reference Trastour134), as well as a predictive biomarker when designing new treatment regimes. Patients with VHL-mutated sporadic clear-cell renal carcinomas exhibit high basal HIF-α expression and subsequently increased expression of VEGF and platelet-derived growth factor (PDGF). To date, the small-molecule tyrosine kinase inhibitors sunitinib and sorafenib that block VEGF and PDGF signalling have been the only agents shown to stabilise the disease, although increases in overall survival are disappointingly low (Refs Reference Bastien69, Reference Motzer135, Reference Motzer136, Reference Ratain137). Both sunitinib and sorafenib have been approved for the treatment of renal cell carcinoma, where sunitinib is currently recommended as a first-line treatment and sorafenib as a second-line treatment option for people with advanced metastatic renal cell carcinoma (Refs Reference Bastien69, Reference O'Brien, Russo and Motzer138). Clinical trials are under way to investigate the efficacy of these drugs in combination with conventional treatments or other small-molecule inhibitors and antibody therapies. Interestingly, a recent cell-based assay approach using renal carcinoma cells has identified STF-62247, an agent that functions to selectively induce cytotoxic and antitumour effects in pVHL-deficient renal carcinoma cells by inducing autophagy (Ref. Reference Turcotte139). The identification of STF-62247 illustrates the possibility of developing therapeutic agents that specifically target pVHL-deficient renal cancers (with high basal HIF-α expression), which account for around 75% of renal cell carcinomas (Ref. Reference Turcotte139). Moreover, a pilot synthetic lethal screen has identified that inhibition of kinases such as CDK4/6 enhanced growth inhibition of pVHL-deficient renal cell carcinomas compared with matched pVHL-proficient renal cell carcinomas (Ref. Reference Bommi-Reddy140), further highlighting those tractable targets for therapeutic intervention in renal cell carcinoma. It will be of particular interest to establish whether HIF inhibitors when used either alone or in combination with these other agents can provide a better therapeutic outcome for patients with advanced metastatic renal cell carcinoma.
Imaging hypoxia as a clinical tool
Effective imaging of hypoxia is important for cancer detection and diagnosis, assessment of therapy, as well as drug development. Imaging also provides a prognostic basis by which the effects of tumour hypoxia could be evaluated in personalised cancer treatment. For example, patients could be selected for hypoxia and high HIF-1α protein levels in their tumours, in order to enter them into clinical trials involving HIF inhibitors.
Several direct and indirect methods for measuring hypoxia have been developed. Direct pO2 measurement can be performed by an Eppendorf probe (a polarographic needle microelectrode) by direct insertion into tissues (Refs Reference Doll141, Reference Movsas142). Exogenous markers of hypoxia (such as pimonidazole and EF5) provide a more reliable measurement than using Eppendorf probes and can be used to detect the distribution of hypoxia in a small fraction of tumour (Refs Reference Kaanders143, Reference Evans144, Reference Raleigh145). Positron emission tomography (PET) tracers such as [18F]fluoromisonidazole have also been used to measure hypoxia throughout the body (Refs Reference Brahme146, Reference Padhani147, Reference Rajendran and Krohn148). PET imaging is sensitive and has a spatial and temporal resolution suitable for accessing the heterogeneity of local pO2. This technique was shown to have the ability to predict outcomes from radiation therapy by imaging hypoxic tissues (Refs Reference Gagel149, Reference Rajendran150, Reference Thorwarth151, Reference Troost152). Several magnetic resonance (MR)-based imaging (MRI) and spectroscopy strategies have been developed to assess tumour hypoxia directly or indirectly – namely 19F-MRI and spectroscopy, high molecular weight dynamic contrast-enhanced MRI, electron paramagnetic resonance imaging and electron paramagnetic resonance oximetry. These methods can reliably and precisely reveal heterogeneity of oxygen distribution within tissues. As well as locating tumour and assessing treatment, MRI-based techniques can detect detailed metabolic and physiological information and PET imaging can reveal biochemical characteristics of the tumour, including metastases. With the preclinical development of many new HIF inhibitors being pursued currently, one challenge will be to define robust preclinical noninvasive imaging endpoints that are not only consistent with measurable effects on the HIF pathway in vivo, but that also directly relate to the mechanism of action and provide clear therapeutic insight with respect to the hypoxic tumour compartment.
Conclusions
Hypoxic tumours are usually resistant to killing by radiotherapy and conventional chemotherapies, rendering them highly aggressive and metastatic. Oxygen homeostasis in cells and the response to hypoxic stress is largely mediated by the HIF pathway. However, dysregulation of the HIF pathway occurs in many human cancers and usually correlates with a poor prognostic outcome using conventional treatments. Therefore, targeting the HIF pathway provides an attractive strategy to treat hypoxic and highly angiogenic tumours. The combination of HIF inhibitors with existing treatments or new targeted therapies may prove to be useful clinically.
However, the development of appropriate imaging strategies to accurately measure hypoxia in tumours along with the identification of suitable biomarker endpoints will accelerate the translation of new therapeutic combinations into the clinic as well as provide prognostic information that helps tailor specific treatments to different cancer types. HIF inhibitors are in preclinical and clinical development and these offer a novel and attractive approach for the treatment of solid tumours.
Acknowledgements and funding
We thank Dr Jan Bussink (Department of Radiation Oncology 874, Radboud University Nijmegen, The Netherlands) for kindly providing the data presented in Figure 1. We also thank Dr Yann Jamin (Institute of Cancer Research, Sutton, Surrey, UK) for his advice on imaging, and Prof. Patrick Maxwell (University College London, UK) for critical review of our manuscript. E.P. is funded by Cancer Research UK project grant C7358/A9958. We also thank the reviewers for their insightful comments and helpful suggestions.






