Introduction
Part one of this series focused on high-throughput nucleotide sequencing and gene expression analysis. However, there are a multitude of other modern high-throughput molecular biology techniques, each with its own bioinformatics methodology and challenges. Many of the principles from sequencing and transcriptional analysis can be applied to the other molecular biology platforms. Here, we discuss other common high-throughput techniques, and briefly summarise the approach to analysis for each.
Comparative genome hybridisation arrays
These arrays are used to detect copy number changes (or copy number variations) in the genome (i.e. deletions or amplifications of specific regions). This is accomplished using microarrays with thousands of DNA probes (small fragments of complementary DNA) designed to analyse the presence or absence of sequential regions along the genome. Usually, DNA extracted from a test sample and that of a reference sample (for example, a tumour vs a normal blood sample in the same patient) are compared on a single array using a two-colour system. The signal intensity at each probe is equivalent to a relative copy number for that small region of the genome.
Just as in gene expression analysis, signals on the array must be normalised and filtered, both internally on an individual chip and across chips, to properly compare samples in an experiment. Copy number variation ‘calls’ involve using probe expression and that of adjacent probes in the genome to identify regions where there is allelic loss or amplification.Reference Wineinger, Kennedy, Erickson, Wojczynski, Bruder and Tiwari1 Similar to gene expression arrays, comparative genome hybridisation array data must be normalised, filtered and analysed critically in order to determine if variation in probe expression is due to normal variation or experimental error, or truly representative of copy number. The resolution of the array depends on how much ‘space’ is between each probe; for example, if a copy number variation occurs in between two regions of probed DNA, it will not be detected. Current comparative genome hybridisation arrays have a resolution of between 100 and 10 000 base pairs, depending on the array.
A recent report by Morris and colleagues used comparative genome hybridisation arrays to describe common copy number variations in head and neck squamous cell carcinoma (SCC).Reference Morris, Taylor, Bivona, Gong, Eng and Brennan2 In this study, which acts as an example of the application of comparative genome hybridisation arrays in head and neck cancer, several alterations in the EGFR/PI3K pathway were identified, including novel microdeletions in the PTPRS gene.
Single nucleotide polymorphism arrays
Single nucleotide polymorphism arrays are microarrays that contain probes specifically examining the presence or absence of single nucleotide polymorphisms, which are single base pair variations in the human genome that are known to occur with regular frequency in the human genome. There are approximately 20 million described single nucleotide polymorphisms,Reference Abecasis, Altshuler, Auton, Brooks and Durbin3 and current arrays can interrogate approximately 1 million of these on a single chip.
Single nucleotide polymorphism arrays are used to genotype an individual for these polymorphisms. The data can be used to carry out linkage analysis and genome-wide association studies. The concept underlying these studies is that inherited polymorphisms that are near a germline mutation or disease-related gene will be inherited in a Mendelian pattern. The relative intensity of signals for polymorphisms on the array can also be used to estimate copy number variations and structural variants along the genome.Reference Wineinger, Kennedy, Erickson, Wojczynski, Bruder and Tiwari1 When utilised in this fashion, bioinformatics approaches to call copy number variations resemble those used with comparative genome hybridisation arrays, with normalisation steps to determine signal thresholds that correspond to copy number.
When using single nucleotide polymorphism data to evaluate copy number variations in cancer studies, it is most appropriate to compare single nucleotide polymorphism data derived from tumour DNA (preferably enriched for tumour cells via pathological assessment and/or laser-capture microdissection) with baseline single nucleotide polymorphism expression in a normal tissue DNA reference from the same patient. A review by Chen and Chen summarises several studies of copy number variations in head and neck SCC, and further discusses the utility of single nucleotide polymorphism arrays.Reference Chen and Chen4
MicroRNA expression arrays
MicroRNAs are small, non-coding fragments of RNA that regulate gene expression, often silencing genes by binding to specific messenger RNA (mRNA) transcripts leading to their degradation.Reference Chen and Rajewsky5 Disruption of microRNA regulation has been implicated in a multitude of diseases.
Global expression of microRNA can be evaluated with microarrays designed to probe hundreds of known microRNAs at a time. Analysis of these arrays is essentially equivalent to gene expression arrays, except that probes correspond to known microRNAs instead of mRNA transcripts.
Examples of microRNA evaluation in head and neck SCC include studies from our own institution (Childs et al.Reference Childs, Fazzari, Kung, Kawachi, Brandwein-Gensler and McLemore6 and Harris et al.Reference Harris, Jimenez, Kawachi, Fan, Chen and Belbin7). The latter study used a unique bioinformatics approach. The ratio of tumour versus normal microRNA expression was calculated for each sample, and then a rank consistency score was used to identify which microRNAs were consistently over- or under-expressed among samples. Using this process, miR-375 was identified as the most consistently decreased transcript among head and neck SCC tumours, and low levels of miR-375 were associated with poor survival.Reference Harris, Jimenez, Kawachi, Fan, Chen and Belbin7
DNA methylation arrays
Another major source of epigenetic regulation is via methylation of DNA. The addition of a methyl group to the 5’ region of cytosines found in gene promoter regions typically causes a reduction of gene expression of the associated gene, and modifications are often found at clusters of CpG dinucleotides (commonly referred to as ‘CpG islands’).Reference Jaenisch and Bird8
Currently, over 450 000 genome-wide methylation events can be evaluated with methylation arrays such as the Illumina© Infinium HumanMethylation450 BeadChip©. Sample DNA is treated with bisulphite, which converts cytosine bases to uracil, but does not change methylated cytosine. Arrays are constructed with probes that are specific for known CpG sites, and complementary probes contain both the cytosine and uracil versions of each site. Therefore, the methylation status of each CpG site can be evaluated by measuring the hybridisation relative to each complementary probe pair. Bioinformatics methods of analysis are thus similar to comparative genome hybridisation arrays or single nucleotide polymorphism arrays.
As a recent example, a study conducted at our own institution examined DNA methylation events in 118 head and neck SCC tumours, and demonstrated differential methylation events that were unique to the subsite of the tumour and human papilloma virus (HPV) status.Reference Lleras, Smith, Adrien, Schlecht, Burk and Harris9
Proteomics
This article has touched on methodologies and bioinformatics in genomics and epigenetics. Proteomics, the comprehensive evaluation of protein expression, structure, modification and function in biological systems, deserves brief mention here.
High-throughput protein analysis techniques include immunohistochemistry (e.g. tissue microarrays), immunoblotting (e.g. enzyme-linked immunosorbent assays, reverse phase protein arrays), and high-throughput techniques using various chromatographic methods combined with mass spectrometry. Detailed review of these methods is beyond the scope of this review.
Bioinformatics approaches focus on the expression, activation and quantification of proteins; these aspects are analysed to delineate protein networks and signalling pathways, in a manner similar to gene expression. Work by Altelaar and colleagues describes some modern proteomics techniques.Reference Altelaar, Munoz and Heck10
Integrative analyses of high-throughput technology data
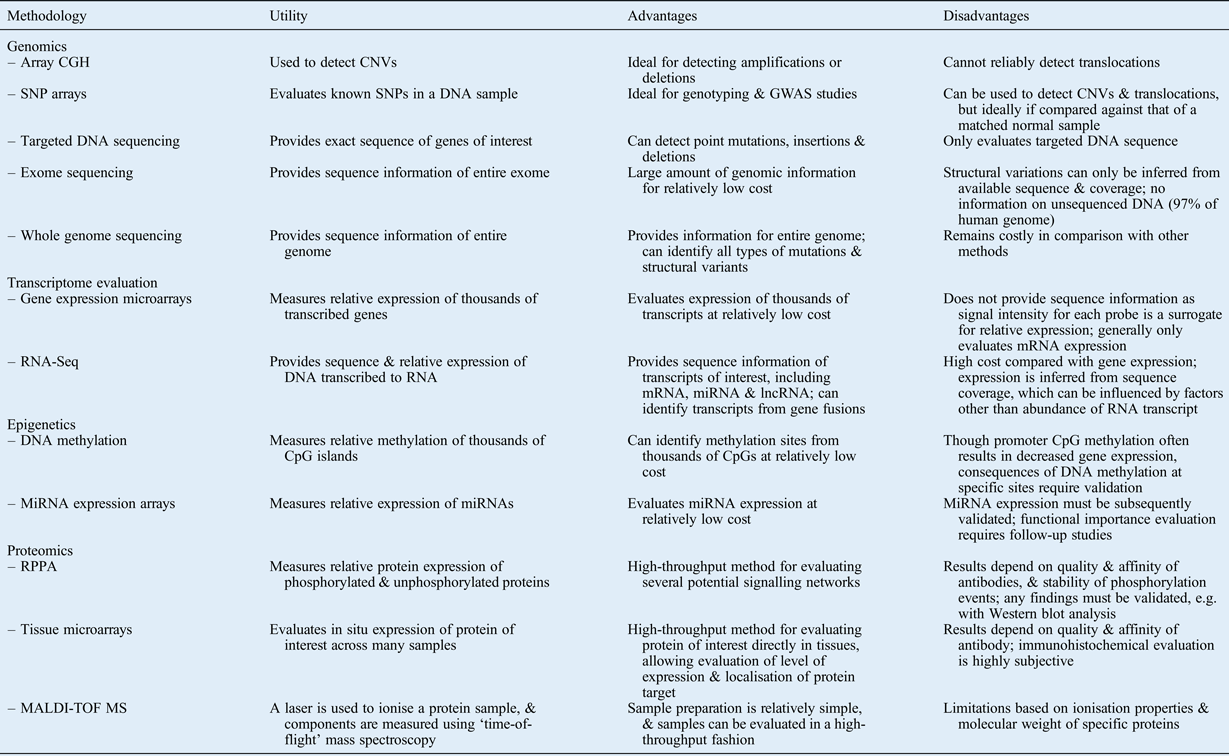
In this review, we have summarised modern comprehensive approaches for evaluating the following: DNA structural changes; sequence alterations; gene expression levels; mechanisms of epigenetic regulation; and protein expression, activation and modulation. Table I summarises several of these methods, describing the utility, and advantages and disadvantages, of each. Note that this list is not comprehensive, as several other methods and variations on the listed methods exist.
Table I Summary of selected high-throughput methodologies: utility, advantages and disadvantages

CGH = comparative genome hybridisation; CNV = copy number variation; SNP = single nucleotide polymorphism; GWAS = genome-wide association study; mRNA = messenger RNA; miRNA = microRNA; lncRNA = long non-coding RNA; RPPA = reverse phase protein arrays; MALDI-TOF MS = matrix-assisted laser desorption/ionisation time-of-flight mass spectroscopy
The bioinformatics approaches used to glean information from each of these platforms individually is complex; however, a greater challenge is to develop methods to integrate these data appropriately, in order to gain a comprehensive understanding of the genetic and molecular underpinnings of human disease. Arguably, the widest applications of integrated analytic approaches have been in the field of cancer biology.
The Cancer Genome Atlas project embodies the modern comprehensive approach to understanding human cancer. The project is an initiative in the USA, supported jointly by the National Human Genome Research Institute and the National Cancer Institute, which aims to comprehensively profile the genetic and epigenetic alterations present in several human cancers. The project studies on glioblastoma, ovarian cancer, colorectal cancer, lung SCC, breast cancer and endometrial cancer have been completed.11–15 The evaluation of several more cancers is underway. The project investigating head and neck SCC has been completed, and the first report is currently in preparation. Information regarding The Cancer Genome Atlas can be found at the project's website.16 The project data for head and neck SCC are now publically available.
Methods for the integration of multiple high-throughput technology research platforms are actively being developed. Several programs have been designed to facilitate interfacing and amalgamation of these types of data. A recent review by Berger et al. lists several available tools.Reference Berger, Peng and Singh17 The methods are not standardised and the approach depends largely on the data available and the experimental questions being asked.
In head and neck SCC, several groups have reported findings gleaned from data combined from two or more high-throughput platforms. A recent study used single nucleotide polymorphism arrays to determine copy number variations, and used gene expression arrays to evaluate 17 tumours without lymph node metastases and 20 lymph node metastases.Reference Xu, Wang, Liu, Zhang, Fan and Upton18 First, differentially expressed genes between the two groups were selected, with a false discovery rate of less than 5 per cent, leaving 1988 transcripts. The data from single nucleotide polymorphism analysis were then used to filter this list by selecting genes whose relative expression was correlated with regions of copy number loss or gain. This left a 95-transcript signature, which was then evaluated on an independent dataset of 133 patients. In a multivariate analysis, the signature was associated with decreases in overall survival and disease-specific survival. Furthermore, amplified genes in the signature were targeted in an in vitro system with a small interfering RNA library, which led to consistent growth suppression in multiple head and neck SCC cell lines.Reference Xu, Wang, Liu, Zhang, Fan and Upton18
In a study from our own institution, DNA methylation arrays were performed on 118 head and neck SCC tumour–normal pairs.Reference Lleras, Smith, Adrien, Schlecht, Burk and Harris9 Differentially methylated genes could distinguish between head and neck SCC and normal tissue. Methylation events specific to tumour subsite and to HPV status were also identified. The expression levels of three highly methylated genes seen in head and neck SCC tumors, ZNF132, ZNF154, and UCHL-1, were then evaluated in an available corresponding gene expression dataset, and these genes were found to be consistently down-regulated.Reference Lleras, Smith, Adrien, Schlecht, Burk and Harris9
Combining data from two platforms is challenging, but a truly integrated approach involves analysing gene networks from data on multiple platforms, exploring genetic alterations, epigenetic regulation and gene transcription in a comprehensive manner. The field of systems biology, as applied to cancer research, aims to use a holistic approach, simultaneously incorporating data from several disciplines in order to evaluate cell signalling networks and model systems, so as to better understand the determinants of cancer cell function, behaviour and phenotype.
Our understanding of how best to integrate multi-platform datasets is in its infancy, but current studies are forging ahead. One recent study by Pickering et al. used whole exome sequencing, single nucleotide polymorphism analysis, DNA methylation arrays, microRNA expression and gene expression arrays to evaluate 38 patients with oral SCC.Reference Pickering, Zhang, Yoo, Bengtsson, Moorthy and Neskey19 As collective events were considered from these platforms, it appeared that specific cell signalling pathways were consistently altered in distinct subsets of patients. It was noted that very few genes were altered in the majority of samples, even when multiple genomic and epigenomic events were considered. When multiple genomic or epigenomic events were assessed in related genes, an accumulation of disruptions in dominant pathways was revealed, including alterations in TP53-related pathways, notch signalling, cell cycle regulators and the EGFR/PI3K mitogenic signalling pathways. Furthermore, comprehensive evaluation across the patient set showed amplifications or activating mutations in oncogenes that could be targeted with existing molecular therapeutic agents in the majority of patients, though any single event was largely under-represented in the cohort.Reference Pickering, Zhang, Yoo, Bengtsson, Moorthy and Neskey19
The Cancer Genome Atlas project is currently underway and, consistent with previous reports for other tumour sites, the head and neck SCC study should provide a comprehensive evaluation of this disease on a large cohort of patients. As the data are publically available, mining this resource using several bioinformatics strategies should greatly advance our understanding of head and neck SCC, and lead to improved therapeutic strategies.
Several open-source databases have been created to make the ever-increasing amounts of biological data available to the public via the internet. Table II presents several online resources that the authors have found useful for a wide range of data analyses, including genome evaluation, gene expression and cell signalling pathway analysis databases. These tools are invaluable for understanding results from research using high-throughput technology, and for exploring relationships between findings in both single- and multi-platform analyses.
Table II Publically available genomics, epigenetics and molecular biology databases
3D = three-dimensional; UCSC = University of California Santa Cruz; MIAME = Minimum Information About a Microarray Experiment standard; miRNA = microRNA
Conclusion
The goal of this review was to introduce the reader to current high-throughput assays available for translational research in otolaryngology, with a focus on the analytical principles of the bioinformatics methods used to understand data derived in such studies. Progress in treating human disease in general will require close collaboration with experts in bioinformatics. Improved understanding of these concepts by clinicians and physician-scientists in our field will advance diagnosis and therapy for diseases of the head and neck.