


Introduction
Familial syndromes involving polyposis and colon cancer underlie 5% of colon cancer cases. Medical surveillance, and in some cases prophylactic surgery, can reduce morbidity and mortality in affected individuals (Gala & Chung, Reference Gala and Chung2011). While clinical data are indispensable to the diagnostic workup, a definitive diagnosis normally rests on genetic testing. In a substantial number of patients presenting with familial colorectal cancer no underlying genetic defect is detected (Lindor, Reference Lindor2009).
Hereditary mixed polyposis syndrome (HMPS) is a genetically heterogeneous, autosomal dominant condition characterized by adenomatous, hyperplastic, and in some cases, juvenile polyps (Whitelaw et al., Reference Whitelaw, Murday, Tomlinson, Thomas, Cottrell, Ginsberg, Bukofzer, Hodgson, Skudowitz, Jass, Talbot, Northover, Bodmer and Solomon1997). BMPR1A and MUTYH mutations account for a small number of cases (Boparai et al., Reference Boparai, Dekker, Van Eeden, Polak, Bartelsman, Mathus-Vliegen, Keller and van Noesel2008; Cheah et al., Reference Cheah, Wong, Chau, Loi, Lim, Lim, Koh and Eu2009; O'Riordan et al., Reference O'Riordan, O'Donoghue, Green, Keegan, Hawkes, Payne, Sheahan and Winter2010), but the underlying genetic defects remain unknown in the majority. Recently an unusual genetic alteration, a 40 kb duplication in the 3′ end of the SCG gene upstream of GREM1, was found among a series of Ashkenazi Jewish HMPS families (Jaeger et al., Reference Jaeger, Leedham, Lewis, Segditsas, Becker, Cuadrado, Davis, Kaur, Heinimann, Howarth, East, Taylor, Thomas and Tomlinson2012). This duplication causes overexpression of GREM1, which is a negative regulator of BMP signalling. The pathogenesis of polyps in HMPS likely overlaps with juvenile polyposis syndromes caused by inactivating mutations in genes of the BMP pathway.
There are limited data on the prevalence of GREM1 mutations in colon cancer families. Based on specific testing for this rearrangement it appears to be extremely rare among unselected colon cancer cases (Jaeger et al., Reference Jaeger, Leedham, Lewis, Segditsas, Becker, Cuadrado, Davis, Kaur, Heinimann, Howarth, East, Taylor, Thomas and Tomlinson2012) and among non-Ashkenazi patients with multiple hyperplastic polyps (Clendenning et al., Reference Clendenning, Young, Walsh, Woodall, Arnold, Jenkins, Win, Hopper, Sweet, Gallinger, Rosty, Parry and Buchanan2013). This gene has not previously been included in targeted panels for hereditary polyposis or Lynch syndrome. Even in more comprehensive tests, such as whole-exome sequencing, the usual analysis focuses on point mutations and small indels, and would miss a 40 kb duplication. Analysis of GREM1 for upstream duplications and other activating mutations among a large set of polyposis and colon cancer cases will be required in order to determine its attributable risk among patients with these phenotypes.
With the identification of the GREM1 duplication, a more complete and less biased description of the spectrum of malignant and non-malignant clinical features in Ashkenazi HMPS can be compiled by examining non-proband members of families who test positive for the mutation. We conducted a comprehensive clinical evaluation of a large Ashkenazi Jewish family presenting with mixed polyposis and early-onset colon cancer. Extensive genetic testing revealed the GREM1 upstream duplication. Our findings further support the association between GREM1 and HMPS, and help define phenotypic features of this condition.
Materials and methods
Subjects
A large HMPS kindred was ascertained through a 21-year-old proband with metastatic colon cancer and his 23-year-old sister who presented for genetic counselling. Written informed consent was obtained from 18 family members who provided blood or saliva samples. Among these subjects seven underwent a complete examination by a medical geneticist, full body skin examination by a dermatologist, and a full ophthalmologic exam. Medical records and pathology reports were sought from all affected family members and were used to determine polyp counts and histopathology. This work was reviewed and approved by the Yale University Institutional Review Board.
Genetic testing
Whole-exome sequencing was performed by the Yale Center for Genome Analysis following procedures available at http://ycga.yale.edu/about/writeups.aspx. Briefly, sheared genomic DNA was captured on a Nimblegen, SeqCap EZ Exome version 2 following the manufacturer's protocols (Roche/NimbleGen). Captured libraries were sequenced on the Illumina GAII as 75-bp single-end reads. Image analysis and base calling was performed by Illumina pipeline versions 1·3 and 1·4 with default parameters. Variant calling used an integrated pipeline that employed SAMtools and BWA.
The 40 kb duplication upstream of GREM1 was detected by PCR using the primers described previously (Jaeger et al., Reference Jaeger, Leedham, Lewis, Segditsas, Becker, Cuadrado, Davis, Kaur, Heinimann, Howarth, East, Taylor, Thomas and Tomlinson2012). Each PCR reaction contained 7 μl ddH2O, 2·5 μl PC2 buffer, 2·5 μl dNTP, 1·25 μl DMSO, 10 μl 5M betaine, 0·2 μl of a mixture of Klentaq (15 parts) and Pfu DNA polymerase (1 part), and 0·5 μl each of forward and reverse primers, both at 10 μM. PCR was carried out in an Eppendorf Mastercycler using cycling conditions: 96 °C (30 sec), 55 °C (30 sec) and 72 °C (40 sec) for 35 cycles. Products were visualized on a 3% agarose gel stained with 5% ethidium bromide.
Results
Clinical report
Medical records and pathology reports obtained from 18 family members established a three-generation pedigree with multiple cases of mixed polyposis and/or colorectal cancer (Fig. 1). The 21-year-old proband had a history of one serrated adenoma, one tubulovillous adenoma, one tubular adenoma and eight hyperplastic polyps as well as metastatic colon cancer. His 23-year-old sister was found on colonoscopy to have one tubulovillous adenoma, one tubular adenoma and two hyperplastic colon polyps. She elected to have a total colectomy because of her brother's outcome.
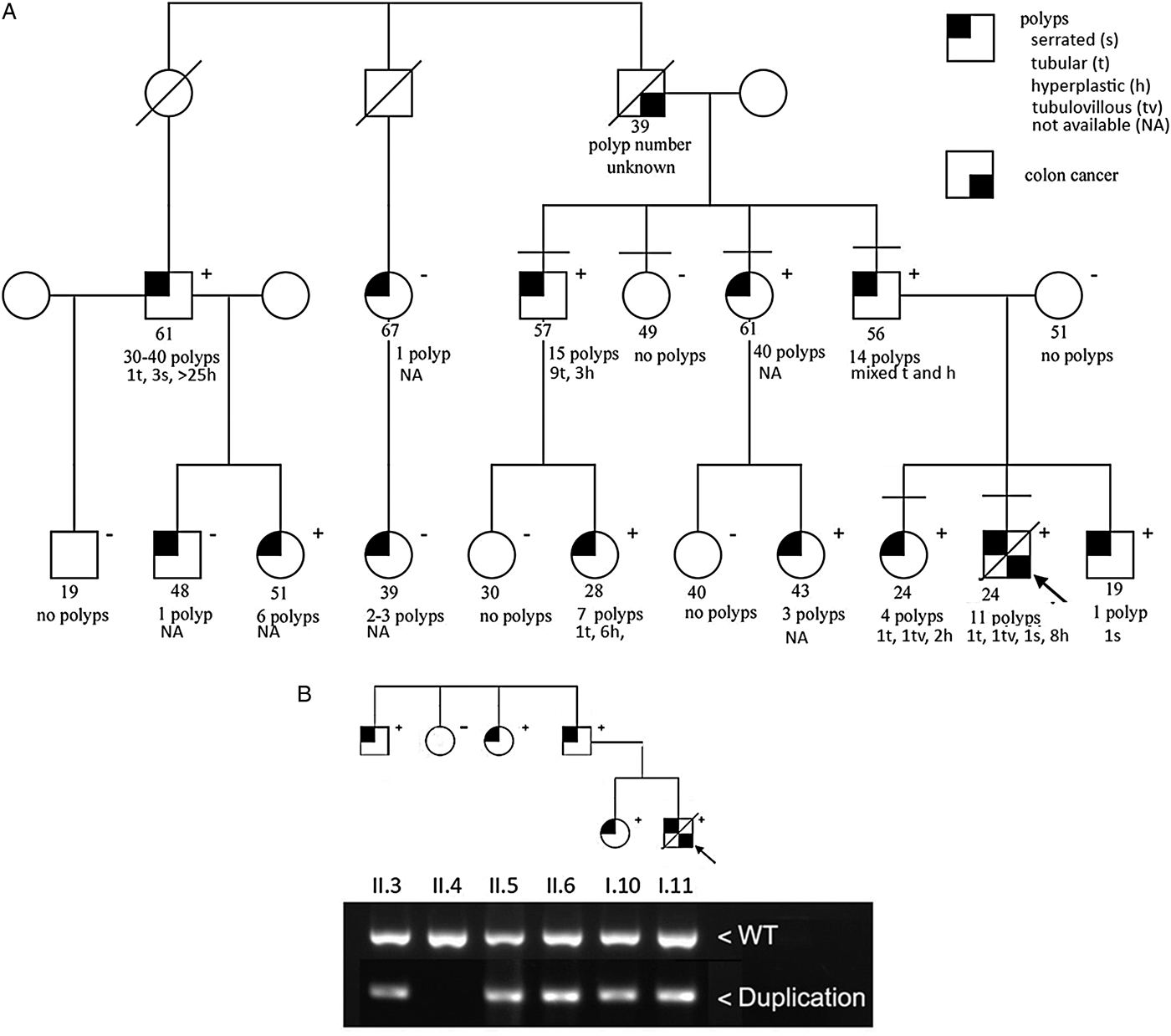
Fig. 1. (a) Pedigree showing age, cancer and polyp phenotype, and presence (+) or absence (−) of a 40 kb duplication upstream from GREM1. Symbols with horizontal bars above them indicate family members also shown in (b). (b) PCR product from wild type (WT) region upstream of GREM1 and an aberrant product produced with PCR primers that flank a duplication breakpoint (Jaeger et al., Reference Jaeger, Woodford-Richens, Lockett, Rowan, Sawyer, Heinimann, Rozen, Murday, Whitelaw, Ginsberg, Atkin, Lynch, Southey, Debinski, Eng, Bodmer, Talbot, Hodgson, Thomas and Tomlinson2003).
Among 18 family members for whom medical records were available, at least one polyp was present in 13, all in the paternal lineage. The proband's mother was unaffected and had no significant family history of colorectal cancer or polyps.
Detailed pathology records obtained from seven individuals indicated that the majority of polyps were tubular or hyperplastic. In addition, there were five serrated polyps among three individuals. Two individuals each had one tubulovillous adenoma. Pathology reports detailing the type(s) of polyps in six other family members were sought but were not available. As part of this study, seven affected family members underwent detailed clinical evaluation. A full examination by a medical geneticist did not reveal any major or minor developmental anomalies. An ophthalmology exam including indirect ophthalmoscopy showed no lesions. A full body skin exam by a dermatologist did not demonstrate any unusual skin findings.
Genetic testing
At the time the family was ascertained, the affected brother had already undergone full sequencing of APC, which showed no mutation, and full sequencing of MUTYH, which showed a variant of unknown significance (P451A) found to be inherited from his unaffected mother. Immunohistochemistry of his colon tumor for MLH1, MSH2 and MSH6 showed no loss of these proteins. Prior to his death he contributed a blood sample for research as part of this study. Whole-exome sequencing identified no mutations in genes known or suspected to be associated with predisposition to colon polyps and colon cancer including APC, AXIN, BMPR1A, CTTNB1, MLH1, MSH2, MLH3, MSH6, PMS1, PMS2, PTEN, SMAD4 and TP53. The previously identified variant in MUTYH was again seen.
During the course of the genetic evaluation the 40 kb duplication upstream of GREM1 was reported in Ashkenazi Jewish families with HMPS (Jaeger et al., Reference Jaeger, Leedham, Lewis, Segditsas, Becker, Cuadrado, Davis, Kaur, Heinimann, Howarth, East, Taylor, Thomas and Tomlinson2012). Published primer sequences were used to test members of the current kindred for the duplication (Fig. 1).
Correlation between GREM1 test results and clinical findings
Among 10 family members testing positive for the GREM1 mutation, polyps were seen in all 10. Detailed pathology was available for seven of these individuals. With the exception of one 19-year-old, who had only one serrated polyp, all others had a mixture of tubular and hyperplastic polyps with several family members also having serrated polyps and tubulovillous polyps. The number of polyps ranged from one in a 19-year-old to 40 in a 61-year-old (mean polyp number 14·1). Notably, multiple polyps were seen in every affected family member over the age of 20.
Among eight family members testing negative, three had one or more polyps (mean polyp number 0·625). No polyps were seen in individuals below the age of 30 and only one individual younger than 40 had any polyps.
Discussion
We performed clinical evaluation and genetic testing of an Ashkenazi Jewish kindred presenting with features of HMPS and found segregation of the 40 kb duplication upstream of GREM1 with polyp formation and colon cancer. This study confirms results of the single previous research study showing an association between the GREM1 duplication and HMPS in Ashkenazi Jewish patients (Jaeger et al., Reference Jaeger, Leedham, Lewis, Segditsas, Becker, Cuadrado, Davis, Kaur, Heinimann, Howarth, East, Taylor, Thomas and Tomlinson2012).
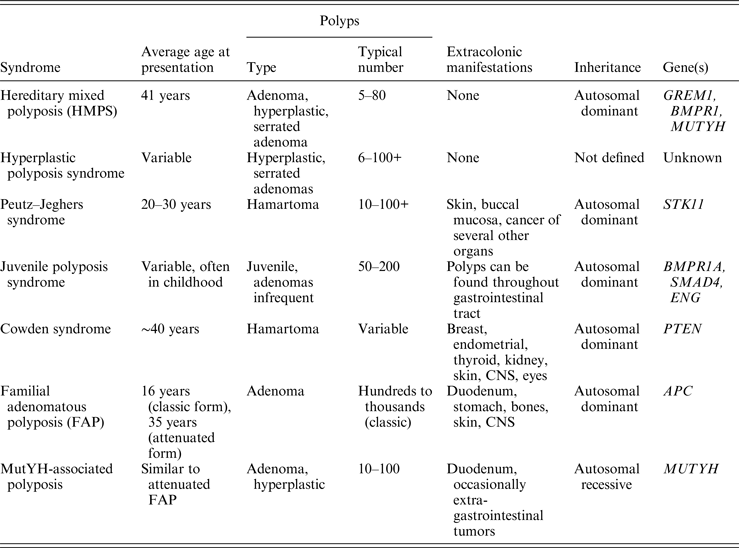
Clinical features of HMPS include polyps confined to the colon and rectum in relatively low numbers, (typically 1–15 and rarely over 50) and colorectal cancer. Inheritance is autosomal dominant, and the average age at presentation is approximately 40 years. Polyp histology includes adenomatous, hyperplastic, and in some cases juvenile polyps (Whitelaw et al., Reference Whitelaw, Murday, Tomlinson, Thomas, Cottrell, Ginsberg, Bukofzer, Hodgson, Skudowitz, Jass, Talbot, Northover, Bodmer and Solomon1997), a clinical constellation distinct from other inherited polyposis or colorectal cancer syndromes (Table 1). Most families with GREM1-linked disease have adenomatous and hyperplastic polyps (Jaeger et al., Reference Jaeger, Woodford-Richens, Lockett, Rowan, Sawyer, Heinimann, Rozen, Murday, Whitelaw, Ginsberg, Atkin, Lynch, Southey, Debinski, Eng, Bodmer, Talbot, Hodgson, Thomas and Tomlinson2003). Juvenile polyps were seen in only one kindred (Whitelaw et al., Reference Whitelaw, Murday, Tomlinson, Thomas, Cottrell, Ginsberg, Bukofzer, Hodgson, Skudowitz, Jass, Talbot, Northover, Bodmer and Solomon1997). Similarly, histologic types in the current family included tubulovillous, hyperplastic, tubular or serrated adenomatous polyps. Juvenile polyps and even partial juvenile histology within a polyp were not reported in any family member. In affected individuals in the current kindred, the average polyp number was 14·1.
Table 1. Clinicopathologic features of the major polyposis syndromes (Patel & Ahnen, Reference Patel and Ahnen2012; Schreibman et al., Reference Schreibman, Baker, Amos and McGarrity2005).
Cancer predisposition syndromes often have skin or eye manifestations (Harbour, Reference Harbour1999; Shah et al., Reference Shah, Boland, Patel, Thrash and Menter2013). In a previous study (Whitelaw et al., Reference Whitelaw, Murday, Tomlinson, Thomas, Cottrell, Ginsberg, Bukofzer, Hodgson, Skudowitz, Jass, Talbot, Northover, Bodmer and Solomon1997) and in our detailed clinical evaluation no manifestations outside the gastrointestinal tract were observed and polyps were restricted to the colon.
Elements of the medical history in this family provide guidance on appropriate genetic counselling and management of this disease. It is not uncommon for patients to report a personal and/or family history of mixed and multiple polyposis. Depending on the exact histology and number of polyps, standard genetic testing to establish a diagnosis and treatment plan may include APC, MYH, PTEN, STK11, SMAD4, BMPRIA and the genes associated with Lynch syndrome. Academic and private clinical laboratories offer large genetic panel testing that includes some or all of the above genes; however, these panels would not typically include the GREM1 upstream duplication. Many laboratories now use whole-exome sequencing to examine large panels of genes, but the GREM1 duplication would not typically be detected by this methodology because analysis of exome data usually focuses on point mutations and small indels. However, targeted examination of sequencing read numbers in the 3′ end of SCG could be used to identify an excess in gene dosage among affected individuals (data not shown).
In Ashkenazi Jewish families with HMPS, GREM1 testing should be the standard of care. A caveat of offering such testing is the limited data available on surveillance and risk reduction. The current study demonstrates that very early-onset colon cancer can occur in GREM1 families. For this reason, we suggest that baseline colonoscopy be offered to at-risk individuals carrying the mutation by age 18 or 5 years before the earliest age of onset, whichever comes first. It would seem reasonable to offer yearly stool guaiac tests and repeat colonoscopies every 5 years to family members with the mutation who have normal colonoscopies. In family members with polyps, a gastroenterologist should guide timing of colonoscopies. Although prophylactic colectomy certainly is an option, the cancer risk and typical age of onset in this syndrome seem similar to attenuated familial adenomatous polyposis, which is usually managed by surveillance (Gala & Chung, Reference Gala and Chung2011).
In summary, our findings further document a relationship between the 40 kb duplication upstream of GREM1 and HMPS in Ashkenazi Jews. Genetic counselling and appropriate molecular testing can lead to successful management of HMPS patients.
Declaration of interest
None.


