


Introduction
First described in 1994 [Reference Murray1], Hendra virus (HeV) (Henipavirus: Paramyxoviridae) continues to cause fatal equine infection and pose a threat of infection in close-contact humans in eastern Australia [Reference Field2–Reference Mahalingam4]. As of 30 April 2019, there have been 103 confirmed or possible equine cases and seven human cases of HeV reported, with case fatality rates around 90% and 60%, respectively [5]. Equine cases occur near-annually; the most recent human case was in 2009, putatively reflecting heightened and targeted awareness and risk mitigation communication by animal and public health authorities. In addition, two canine infections (both associated with equine cases) have been reported [Reference Kirkland6]. Pteropus spp. bats (colloquially known as flying-foxes in Australia) are the natural host of the virus [Reference Halpin, Young and Field7–Reference Field9], with P. alecto and P. conspicillatus shown to be the primary reservoir [Reference Smith10–Reference Edson12].
An effective vaccine for horses is available [13], and vaccination is regarded by animal health authorities as the single most effective means of preventing infection [14]. However, vaccine uptake has been limited, and minimizing contact between horses and flying-foxes remains a primary risk management strategy for many horse owners [Reference Manyweathers15, Reference Manyweathers16]. To be effective, such strategies need to be underpinned by a comprehensive understanding of drivers and dynamics of HeV infection in flying-foxes [Reference Field9, Reference Field17, Reference Plowright18].
Early studies of the ecology of infection in flying-foxes focused on individual animal serology and demonstrated that neutralizing antibodies to HeV were taxonomically and geographically widespread in flying-foxes [Reference Field19]. Various studies have shown a higher antibody prevalence in older flying-foxes, in pregnant and lactating flying-foxes, and in black flying-foxes (Pteropus alecto) [Reference Field19–Reference Breed22]. Later studies sought viral RNA using qRT PCR on pooled urine samples collected under roosting flying-foxes. These studies progressively demonstrated the limited genetic diversity of HeV [Reference Smith23], spatial and temporal heterogeneity in strain occurrence [Reference Smith23], higher probability of detection in black and spectacled flying-foxes [Reference Edson24], and spatiotemporal patterns of excretion [Reference Field9, Reference Paez25]. More recently, qRT PCR approaches have been used in individual animal studies, confirming black flying-foxes as an epidemiologically significant host species, and establishing urine as a primary route of excretion [Reference Goldspink11, Reference Edson12].
While these studies have cumulatively provided information on which to base and refine exposure risk management strategies, the relative infection risk posed by various flying-fox cohorts over time, and the relationship between infection status and antibody status, has remained an enduring knowledge gap. In this longitudinal study, we describe, compare and contrast both serologic and molecular findings in samples from individual bats to better understand HeV infection and immune dynamics in flying-foxes and to better inform equine exposure risk management. Our aim is to identify key epidemiological variables significantly associated with HeV infection in flying-foxes.
Methods
Ethics statement
Fieldwork was approved under the (then) Queensland Department of Agriculture, Fisheries and Forestry Animal Ethics Committee Permit SA 2011/12/375 and the Queensland Department of Environment, Heritage and Protection Scientific Purposes Permits WISP05810609 and WISP14100614. Capture and sampling were undertaken by trained and experienced teams including at least one veterinarian.
Capture and sampling
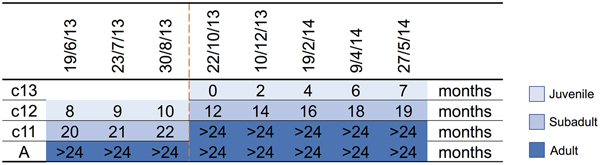
A parkland roost in the town of Boonah in southeast Queensland (27.992°S, 152.681°E) was purposively selected as the sampling location for several related studies because of its apparent permanency, substantial size, species diversity, accessibility and proximity to the Biological Sciences Laboratory (BSL) in Brisbane. Between June 2013 and June 2014, flying-foxes were non-randomly captured and sampled approximately every 2 months as described by Edson et al. [Reference Edson12]. Briefly, animals were captured using mist nets as they returned to roost pre-dawn; each animal was promptly extracted from the net and placed individually in a clean pillow case tied to a horizontal line pending anaesthesia and sample collection. Each bat was subsequently anaesthetised using the inhalation agent isoflurane and medical oxygen [Reference Jonsson26]. Capture and sampling events typically occurred over 7–10 successive days, with the mid-point taken as the sample date for analytical purposes. Species, sex, age class (juvenile, sub-adult, adult), bodyweight (g), forearm length (mm), body condition score (BCS) (1–5) and reproductive status (palpably pregnant, lactating) were recorded. Age class was based on morphometrics and sexual maturity, with juvenile <12 months, sub-adult 12–24 months and adult >24 months [Reference Edson12]. BCS was based on palpated pectoral muscle mass, with 1 = poor, 2 = less than fair, 3 = fair, 4 = greater than fair, 5 = good [Reference Edson12]. Bats were also classified by birth year cohort around the October mid-point of the annual birth pulse [Reference Peel27] (Fig. 1). Thus, we sampled bats born before 30 September 2011 (adults), bats born in the 2011 birth cohort (c11) which were ~20 months (sub-adults) at the beginning of the study and transitioned to adults during the study, bats born in the 2012 birth cohort (c12) which were ~8 months (juvenile) at the beginning of the study and ~19 months (sub-adults) at the end of the study, and bats born in the 2013 birth cohort (c13) which were born during the study period and ~7 months (juvenile) at the end. Adult bats could not be assigned a birth year cohort because of the limitations of ageing techniques based on morphometrics and sexual maturity beyond 24 months. One millilitre of blood was collected from the wing (cephalic) vein of each bat using a 25 G needle and 2 ml syringe, and immediately placed in a serum tube (1.3 ml BD Serum Tube®). Blood was allowed to clot for 6–24 h prior to centrifugation and separation of serum and packed haemocytes. Urine was collected into one or more plain 2 ml cryovials by gentle trans-abdominal compression of the bladder, and samples retained on ice prior to processing. Nasal, oral and rectal swabs (551C Copan® Minitip Flocked Swab) (plus vaginal or preputial swabs when a urine sample was not obtained) were taken, immediately placed in 500 µl of phosphate-buffered saline (PBS) and kept on ice prior to processing. All bats were monitored post-anaesthesia until they regained consciousness, and after a further recovery period of 30–60 min, released at the point of capture.
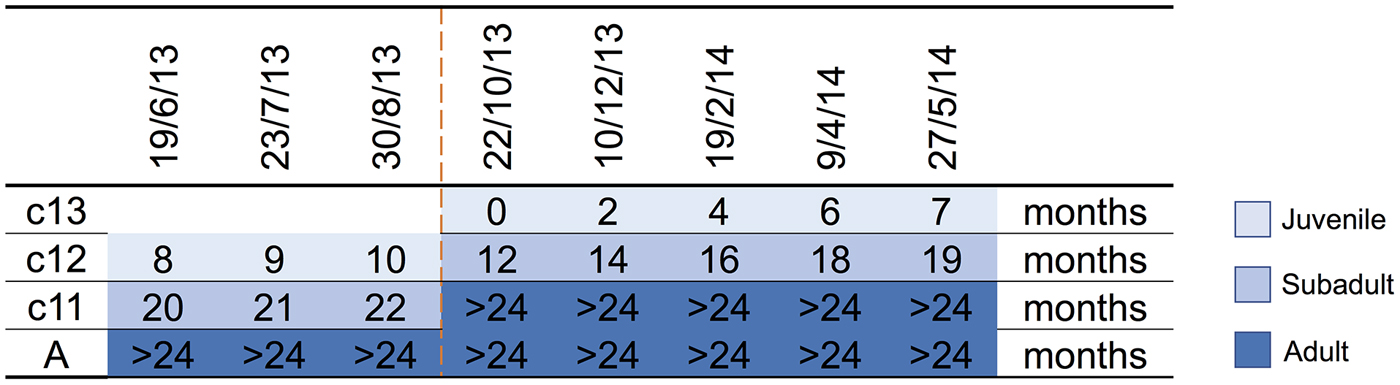
Fig. 1. Birth cohort profiles over the study period, with sampling event midpoints and the age of sampled animal in months indicated. c13 = 2013 birth cohort, c12 = 2012 birth cohort, c11 = 2011 birth cohort, A = adult, and includes animals borne in the 2010 and earlier birth cohorts. The dashed line indicates the October mid-point of the annual birth pulse.
P. alecto were preferentially targeted to facilitate investigation of infection dynamics because of the reported higher HeV detection rate in this species [Reference Field9, Reference Goldspink11, Reference Edson12]. P. poliocephalus and P. scapulatus were included to enable comparative sero-epidemiology.
Sample processing and testing
Serologic testing for anti-HeV antibodies
Serology was performed using a multiplex microsphere technique previously described by Bossart et al. [Reference Bossart28]. Briefly, a pre-determined number of magnetic carboxylated microsphere beads (Fisher Biotec Pty Ltd, Australia) were selected for a protein coupling reaction. The HeV soluble G protein [Reference Bossart29] was coupled to the bead set and mixed with the test sera at a dilution of 1:100. Bound antibody was detected using biotinylated Protein A at 1:500 and biotinylated Protein G at 1:250 (Pierce, USA); the reporter streptavidin-phycoerythrin was added at 1:1000 (Qiagen Pty Ltd, Australia). The beads were analysed using a Luminex MAGPIX system, and results were recorded as median fluorescent intensity (MFI).
The likelihood that an individual MFI value was positive or negative was determined by calculating cut-offs using Bayesian mixture models following Peel et al. [Reference Peel30] and Chowdhury et al. [Reference Chowdhury31]. However, after log-transformation of MFI values, the seronegative and seropositive distributions remained positively and negatively skewed, respectively, indicating that normal distribution models were inappropriate. The mixture model was therefore fitted with a shifted-Gompertz and a Gompertz distribution in R [32]. Conservative species-specific cut-offs were calculated (P. alecto, 1636; P. poliocephalus, 992; P. scapulatus, 1339) so that individuals with MFI values above the cut-off had a probability in excess of 95% of having anti-HeV antibodies. Natural logarithm MFI (lnMFI) values have previously been found to correlate strongly with neutralizing antibody titre [Reference Bossart28, Reference Peel30, Reference Peel33], and are a robust indication of immune dynamics at a population level. Further details, including potential limitations of this assay, are provided as Supplementary information (Text S1, Table S1, Figs S1 and S2).
Molecular testing for HeV RNA
The quantitative RT-PCR data reported in Edson et al. [Reference Edson12] are re-analysed in this paper after pairing with serological data. Briefly, they added 50 µl of urine, serum, packed haemocytes and PBS from nasal, oral and rectal swab samples (plus vaginal or preputial swabs when a urine sample was not obtained) individually to 130 µl of lysis solution (MagMAX, Ambion, Texas, Cat AM8500) to inactivate virus particles and preserve RNA for PCR screening. Total nucleic acid was extracted using a magnetic bead-based nucleic acid extraction kit (MagMAX-96 viral RNA isolation system, Ambion, Texas, Cat AM1836-5) run on a magnetic particle handling system (Kingfisher KF-96, Thermo-Scientific, Finland) according to the manufacturer's instructions. Using the AgPath-ID One-Step RT-PCR Kit (Life Technologies, Melbourne, Australia), 5 µl of nucleic acid extract was added to 20 µl of mastermix. Forward and reverse primers and probe targeting a 69 base pair region on the M gene were used [Reference Smith34]. Positive and negative controls were included in each run. Assays were run on a 7500 Fast Real-Time PCR System (Applied Biosystems) in standard mode for a total of 45 cycles in accordance with the manufacturer's instructions for the mastermix. As described in Edson et al. [Reference Edson12], a subset of individuals (621/1968) had all sample types tested; for the remainder, serum and urine (or vaginal or preputial swabs when urine was not obtained) were used as a screening panel. An individual was considered ‘RNA-positive’ if any sample type yielded a positive PCR result (a Ct value <40), and ‘RNA-negative’ otherwise [Reference Smith34]. However, Edson et al. [Reference Edson12] found that only 9/16 individuals positive on their urine sample were positive on their paired vaginal or preputial swab, suggesting the RNA-negative cohort potentially contains a small number of false-negative results where individuals tested negative on vaginal or preputial swab in the absence of a urine sample (see Discussion). Thus, analysis of the PCR data was undertaken at three levels: animal (individuals with at least one sample type tested), urine (only individuals with a urine sample) and serum (only individuals with a serum sample).
Statistical methods
The effects of sampling date, species, sex, age, BCS, pregnancy and lactation, and their respective interactions, on HeV antibody prevalence, MFI and HeV RNA status were assessed, with the exception that species were excluded from the latter as all RNA detections were in a single species (P. alecto). Data for each variable were subjected to an unbalanced generalised linear model [Reference McCullagh and Nelder35] using GenStat 2013 [36]. The binomial distribution and logit link were adopted for binary data (HeV antibody prevalence and RNA status) and the ln-Normal for MFI data. The initial model included the design variables sampling event midpoint date (sample date), age class, species, sex and the interaction term date.age, with additional models separately adding BCS (as a linear contrast), pregnancy and lactation (as these were correlated). All models were repeated using age cohort instead of age class. The exact binomial method for confidence intervals [Reference Clopper and Pearson37] was adopted for zero prevalence values, as the logit estimates here become unstable. All significance testing was conducted at the 95% level (P < 0.05). Adjusted means (marginal, i.e. weighted by the numbers of observations in each class) and standard errors were estimated for each variable, along with 95% confidence intervals (calculated on the logit scale and back-transformed to percentages). The residual plots for most variables proved to be approximately normal. Those that showed positive skewness and heterogeneous variance were transformed using the natural logarithm (ln).
P. scapulatus were excluded from the serologic analysis as the absence of a clear bimodal distribution of lnMFI values and a suboptimal model fit precluded robust interpretation for this species (Text S1, Table S1, Figs S1 and S2).
Results
A total of 1968 flying-foxes were captured and sampled over the 13-month study period, comprising 1012 P. alecto, 742 P. poliocephalus and 214 P. scapulatus (Fig. S3). PCR results were obtained for all individuals; serology results were obtained for 1906 individuals (967 P. alecto, 734 P. poliocephalus, 205 P. scapulatus). Model outputs and sample sizes of variables significantly associated with HeV RNA detection and antibody detection are presented in Data S1, Table S2 and Table S3, respectively.
RNA prevalence
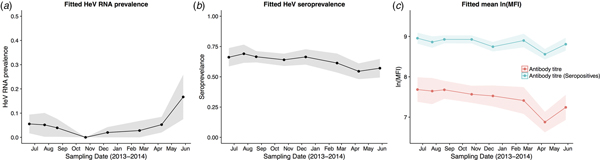
HeV RNA was detected only in P. alecto, with 42 of 1012 individuals (4.2%, 95% CI 3.1–5.6%) positive on one or more sample type, including serum (11/1000), urine (26/558), vaginal and preputial swabs (18/871), rectal swabs (8/318), nasal swabs (3/306) and oral swabs (2/307) [Reference Edson12]. None of the 2013 birth cohort (~0–7 months during the study period, n = 35) tested positive. The youngest bats that tested positive for RNA were sub-adults ~16 months of age (2012 birth cohort), with cohort prevalence increasing to the end of the study when the bats were ~19 months. Limited data points precluded the meaningful interpretation of the 2011 cohort (Fig. 2), but a similar temporal pattern to that seen in sub-adults was evident in the adult cohort. RNA prevalence varied significantly with sample date (peaking mid-year, Fig. 3a) and age class (sub-adults > adults) in the initial model, and with BCS (poor > fair > good), and antibody status (positive > negative) in additional models (Table 1). Of 38 bats that were RNA-positive and had matched antibody data, 34 were antibody-positive and four were antibody-negative. There was no association between RNA prevalence and sex, pregnancy or lactation, although prevalence differed slightly among adult P. alecto that were pregnant (14/195, 7.2%, 95% CI 4.3–11.7), lactating but not pregnant (3/132, 2.3%, 95% CI 0.8–6.5) or were neither pregnant nor lactating (5/96, 5.2%, 95% CI 2.2–11.6). RNA detection in urine was positively associated with sample date and antibody status (positive > negative), but not BCS; RNA detection in serum was positively associated with sample date, age class (sub-adult>adult) and sex (female>male), but not BCS or antibody status. None of the 742 P. poliocephalus or the 214 P. scapulatus yielded a positive PCR result on any sample [Reference Edson12], yielding theoretical upper 95% confidence intervals for infection prevalence for these species of 0.5% and 1.7%, respectively.
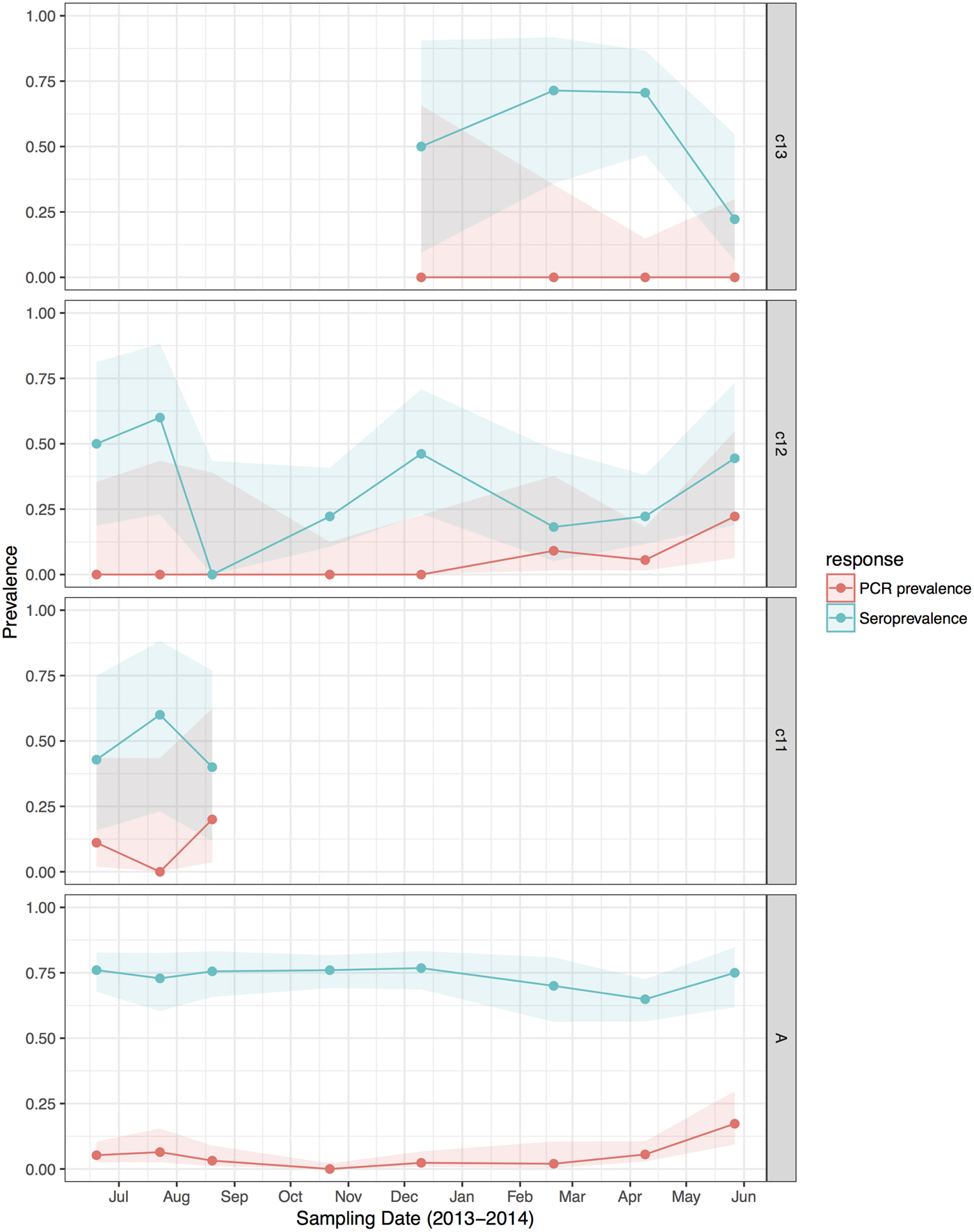
Fig. 2. Hendra virus RNA and antibody prevalence in P. alecto, with juvenile and sub-adult data presented as birth-year cohorts. c13 bats were born in the 2013 birth season and were ~7 months old at the end of the study; c12 bats were born in the 2012 birth season and were ~8 months old at the start of the study and ~19 months old at the end of the study; c11 bats were born in the 2011 birth season and were ~20 months old at the start of the study, and joined the adult (a) cohort during the study period.
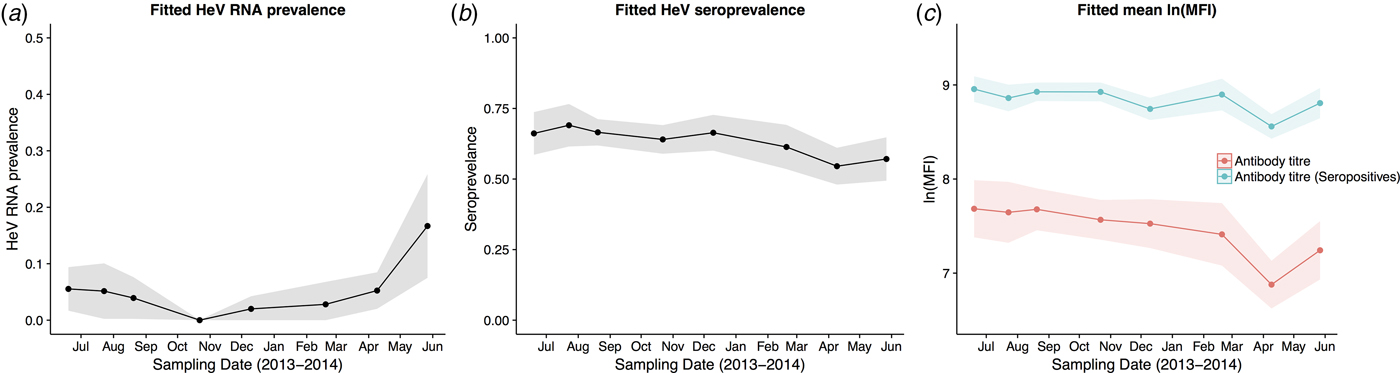
Fig. 3. Model fitted Hendra virus RNA prevalence in P. alecto (a), mean anti-Hendra virus antibody prevalence in P. alecto and P. poliocephalus (b), and mean anti-Hendra virus lnMFI in P. alecto and P. poliocephalus (c).
Table 1. Variables significantly associated with molecular and serological measures of HeV infectiona in wild-caught flying-foxes sampled at Boonah in southeast Queensland in 2012–2013
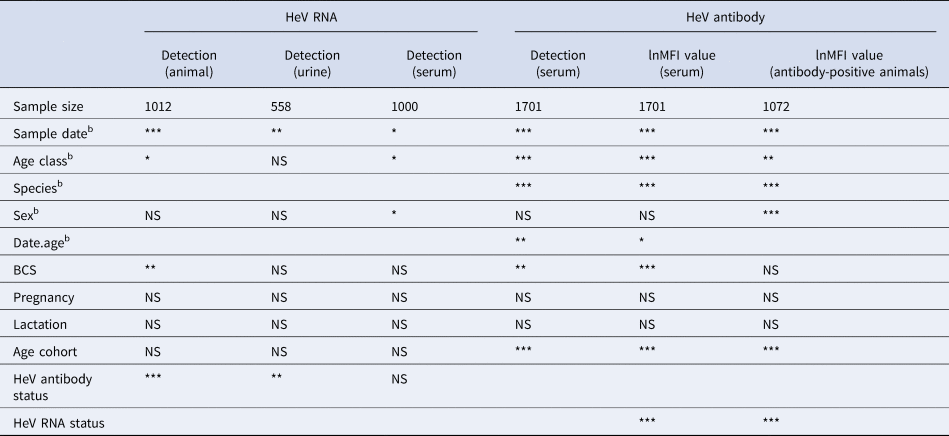
NS, not significant, *<0.05, **<0.01, ***<0.001, blank, association not tested.
a HeV RNA models included data from P. alecto only, as P. poliocephalus did not yield any PCR-positive samples. HeV antibody models included data from both P. alecto and P. poliocephalus samples.
b Initial model variables.
Full results of all models are provided as Supplementary information.
Antibody prevalence
HeV antibody was detected in both P. alecto (651/967) and P. poliocephalus (421/734). Antibody prevalence varied significantly with sample date (declining over the study period) (Fig. 3b), age class (adult > juvenile > sub-adult), species (P. alecto > P. poliocephalus) and the interaction term date.age in the initial model, and with cohort (adult > c13 > c11 > c12) and BCS (good > fair > poor) in additional models (Table 1). For both P. alecto and P. poliocephalus, antibody prevalence was relatively static in adults, but more dynamic in juveniles and sub-adults as illustrated in Figure 2 (P. alecto) and Figure 4 (P. poliocephalus). In the 2013 cohort, prevalence in both species declined by the end of the study when these individuals were approximately 7 months of age. In the 2012 cohort, antibody prevalence in both species was 0% at ~10 months of age (P. alecto 0/80, 0%, 95% CI 0.0–4.6; P. poliocephalus 0/35, 0%, 95% CI 0.00–9.9), then increased across the next two sample dates before fluctuating. Limited data points for the 2011 cohort again precluded meaningful interpretation. There was no association between antibody prevalence and sex, pregnancy or lactation. In adult P. alecto, prevalence did not differ significantly among individuals that were pregnant (143/184, 77.7%, 95% CI 71.2–83.1), lactating but not pregnant (105/129, 81.5%, 95% CI 73.8–87.2) vs. those that were neither pregnant nor lactating (66/92, 71.7%, 95% CI 61.8–79.9). Similarly, antibody prevalence in adult P. poliocephalus did not differ significantly with pregnancy or lactation status, but was lower than in adult P. alecto across all reproductive stages; pregnant (105/165, 63.6%, 95% CI 56.0–70.6.1), lactating but not pregnant (33/57, 57.9%, 95% CI 45.0–69.8), neither pregnant nor lactating (31/62, 50.0%, 95% CI 37.9–62.1).
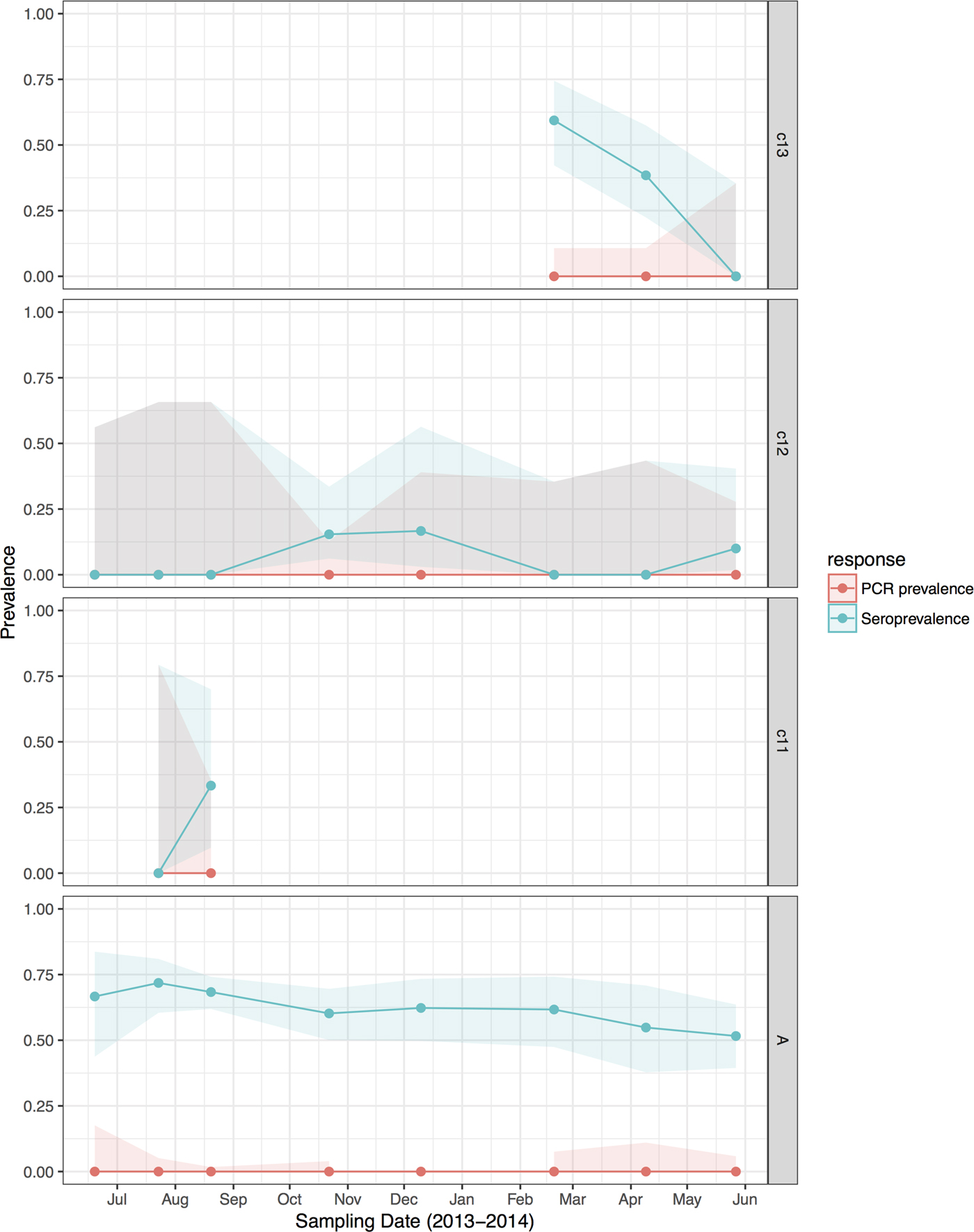
Fig. 4. Hendra virus RNA and antibody prevalence in P. poliocephalus, with juvenile and sub-adult data presented as birth-year cohorts. c13 bats were born in the 2013 birth season and were ~7 months old at the end of the study; c12 bats were born in the 2012 birth season and were ~8 months old at the start of the study and ~19 months old at the end of the study; c11 bats were born in the 2011 birth season and were ~20 months old at the start of the study, and joined the adult (A) cohort during the study period.
Comparative summary viral RNA and antibody prevalence data for P. alecto are presented in Table 2.
Table 2. Comparative Hendra virus molecular and serology findings in 967a P. alecto sampled at Boonah between June 2013 and June 2014
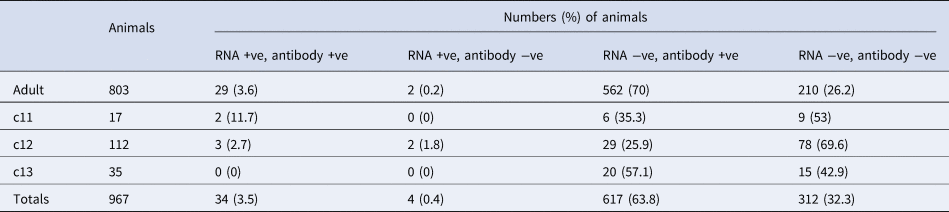
a Nine hundred sixty-seven P. alecto had positive results for both PCR and serology. While 734 P. poliocephalus had positive serology, none had positive PCR results, precluding their inclusion here.
lnMFI value
A significant association was observed between lnMFI and sample date (Table 1, Fig. 3c), age class (adult > juvenile > sub-adult), species (P. alecto > P. poliocephalus) and the interaction term date.age, and with cohort (adult > c13 > c11 > c12), BCS (good > fair > poor) and RNA status (positive > negative) in additional models. In P. alecto, lnMFI values in adults and sub-adults were highest in winter, and in juveniles, highest in summer. There were no significant associations with sex, pregnancy or lactation in adult P. alecto (Fig. S4) or P. poliocephalus (Fig. S5).
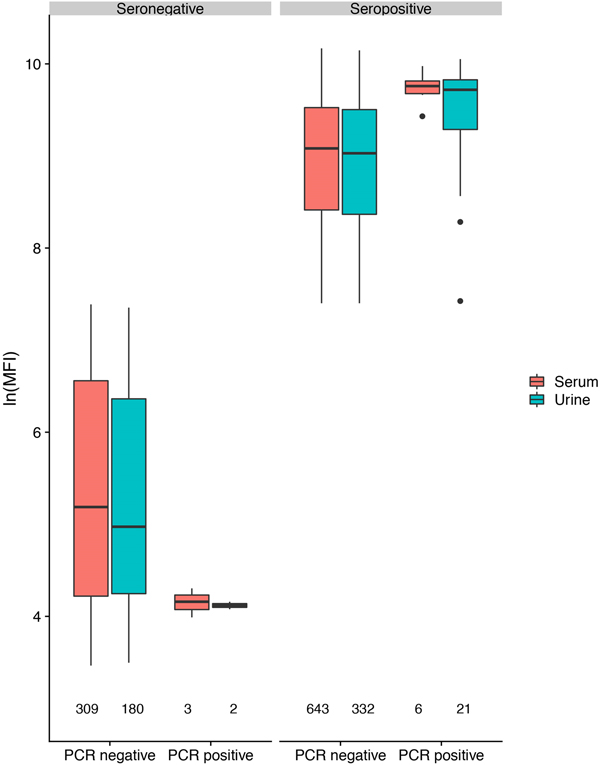
In antibody-positive animals, lnMFI values were significantly associated with sample date, age class (adult>sub-adult>juvenile), species (P. alecto > P. poliocephalus) and sex (female > male) in the initial model, and with cohort (c12 > adult > c13 > c11) and RNA status (positive > negative) in additional models (Table 1, Fig. 3c, Fig. S6). Of 11 P. alecto individuals with HeV RNA detected in serum (seven adult females, three sub-adult females and one sub-adult male), nine also had concurrent antibody data, and seven also had concurrent urine RNA data. Six of the former were antibody-positive, with high lnMFI values (mean lnMFI = 9.7) compared to RNA-negative individuals (mean lnMFI = 7.8) and to the majority of urine RNA-positive individuals (mean lnMFI = 9.4) (Fig. 5). The remaining three serum RNA-positive bats (33%) were antibody-negative, compared to only 8.7% (2/23) of urine RNA-positive bats testing antibody-negative. Of six individuals with urine and serum RNA data plus antibody data, four were concurrently positive across all three tests (all adult females), one was serum RNA-positive and antibody-positive, but urine RNA-negative (adult female), and one was urine and serum RNA-positive, but antibody-negative (sub-adult male).
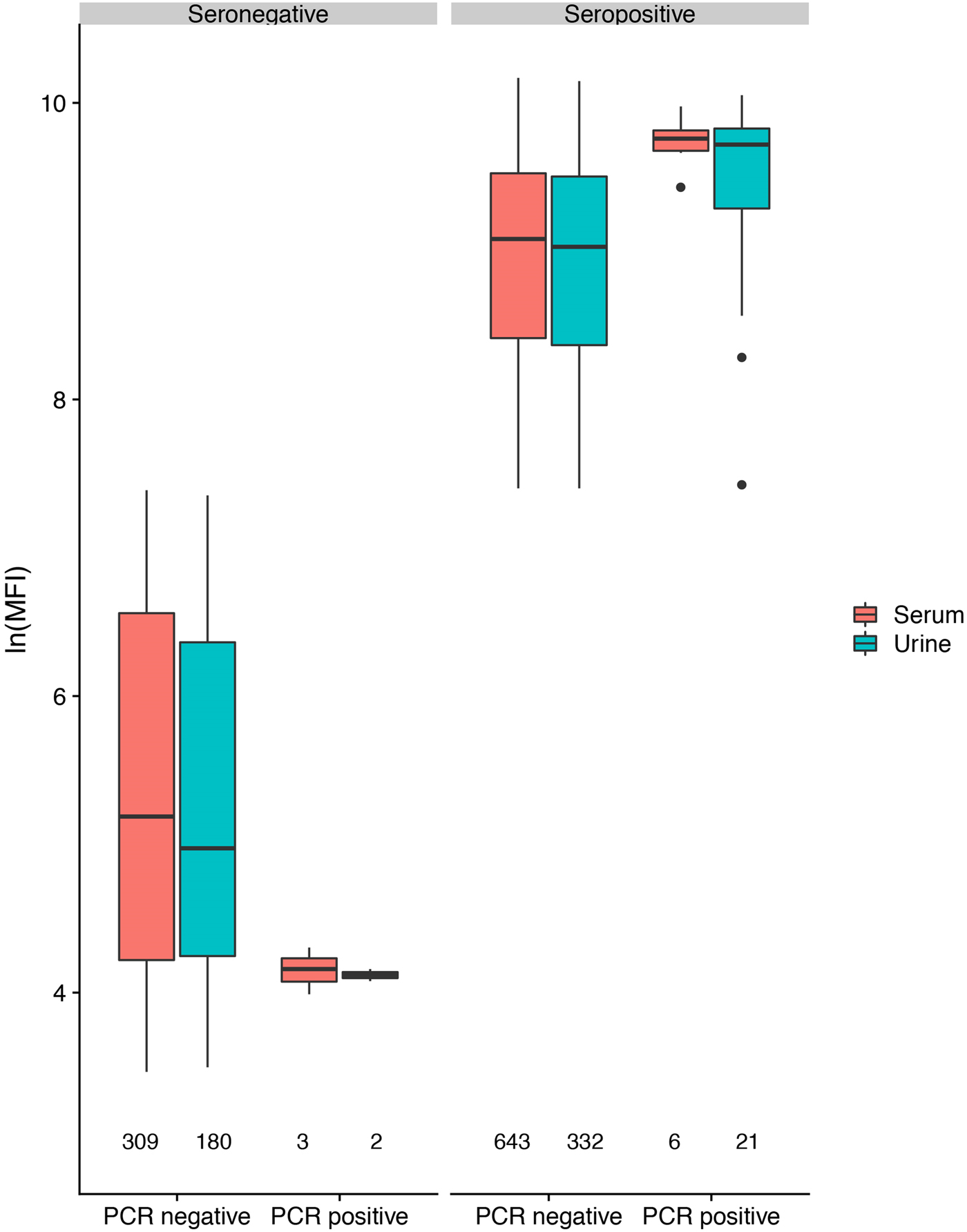
Fig. 5. Hendra virus antibody lnMFI values in P. alecto individuals by HeV RNA detection status in serum or urine.
Discussion
This study sought to identify epidemiological variables significantly associated with HeV infection in a mixed-species colony of free-living flying-foxes over time. With a sample size of 1968 individuals, we sought associations with both RNA status as a measure of current infection, and antibody status and lnMFI values as measures of past infection. To our knowledge, this is the first study to examine concurrent serologic and molecular data of individual flying-foxes, and thus gain an insight into both infection and immune dynamics.
The molecular data used in this analysis has been previously analysed in the context of routes of HeV excretion [Reference Edson12] and showed that HeV RNA detection was limited to P. alecto, with no detection in P. poliocephalus or P. scapulatus. Here, we focus on identified epidemiological associations with HeV RNA detection in P. alecto. We found that RNA detection varied over the year, with infection in the colony more likely in the autumn and winter months. This finding is consistent with those of Field et al. [Reference Field9] across the broader southeast Queensland/northeast NSW region, and with the winter clustering of equine cases in southeast Queensland/northeast NSW [Reference Smith38, Reference Plowright39], although a temporal association with infection (in flying-foxes and horses) appears to be weaker or lacking or beyond this region. The positive association between RNA detection and sub-adults in the 2012 birth cohort is consistent with a seasonal pulse of susceptible individuals as maternal HeV antibodies wane [Reference Epstein40] and suggest sub-adults may play an important role in maintaining HeV infection at a population level. This scenario has previously been hypothesised in relation to HeV [Reference Field and Kung41], and parallels exist in relation to other infectious diseases [Reference Kirkland42–Reference Peel44]. However, given the complex nature of the system, it is likely that additional drivers (e.g. population dynamics, environmental factors) also influence HeV transmission in flying-foxes [Reference Field9, Reference Paez25].
The observed positive association between RNA detection and antibody detection, as well as the higher lnMFI values in RNA-positive individuals, is consistent with seroconversion during infection (or at least before viral clearing), and with seroconversion (and boosted lnMFI values) associated with recurring or persistent infection. Individuals testing positive for HeV RNA in serum were less likely to be concurrently seropositive than individuals testing positive for RNA in urine, but when they were seropositive, they had higher lnMFI values on average. This could suggest that in natural infections, RNA in the peripheral circulation is indicative of very recent infection, with either insufficient time for a detectable IgG response to be mounted (seronegative), or rapid seroconversion to high lnMFI values (seropositive). These findings and interpretations contrast with earlier experimental studies which suggested that infection in serum and urine occurred simultaneously, and that antibody response was unpredictable and not reflective of time since infection [Reference Halpin8]. Notwithstanding, the identified positive association between RNA detection and lnMFI values identified here suggests that recent/current infection provokes a strong IgG response in both sub-adult and adult bats. While the inability of the assay to detect IgM constrains further interpretation, it is plausible that RNA detection in sub-adults more likely represents their first infection, and their lnMFI values a primary response; with increasing age, adults are more likely to have been previously infected. More broadly, it is unclear whether the 26% adult bats that were both RNA-negative and antibody-negative have never been exposed to HeV, or whether they have been exposed but did not develop (or no longer have) detectable antibodies. Regarding the latter, it also remains unknown whether these bats are susceptible to re-infection, or whether primary infection provides lifelong protective immunity against re-infection in the absence of detectable antibodies, as has been suggested in other bat-viral systems [Reference Schuh45].
The statistical association between positive RNA status and lower BCS in the animal-level analysis (but not the urine- or serum-level analyses) is of unknown biological significance. It may simply reflect correlation, with the probability of infection temporally coincident with seasonal weight loss [Reference McMichael46]; alternatively, it could reflect causation, with individuals in poorer body condition more likely to become infected, or at least, more likely to excrete virus. The latter could plausibly include a scenario of recrudescing infection and viral shedding as body condition declines [Reference Plowright21, Reference Wang47]. However, interpretation should consider the possibility of a small number of individuals being misclassified as ‘RNA-negative’ on the basis of a negative vaginal or preputial swab where a urine sample was not available, as Edson et al. [Reference Edson12] found that negative vaginal or preputial swabs under-estimated RNA presence in urine by around 44%. Consistent with our study, McMichael et al. [Reference McMichael48] found a significant association between positive HeV RNA status and lower triglyceride levels, a biomarker which they had previously shown to correlate with lower BCS [Reference McMichael49], but no association in urine-only analyses between HeV status and the majority of other biomarkers strongly correlated with BCS.
The reverse association was found between antibody status and BCS, with animals in poorer condition less likely to have anti-HeV antibodies (and more likely to have lower lnMFI values) than animals in good condition. Immune responses are energetically costly; energy restriction, with or without malnutrition, can result in suppression of the immune system and increased infection and mortality rates [Reference Scrimshaw and SanGiovanni50]. While speculative, our findings may suggest that animals in poorer body condition are less capable of mounting an effective innate and/or humoral immune response, resulting in an increasing likelihood of infection and/or viral excretion. Kessler et al. [Reference Kessler51] suggest that the combined effects of habitat loss and resource provisioning are driving changes in fruit bat feeding ecology and nutrition, and may be associated with increasing risk of viral spillover. Interestingly, the association between poorer body condition and a weaker serologic response (in terms of both serostatus and lnMFI values) was also observed in P. poliocephalus (data not shown) suggesting the same mechanism may be at play, but as previously stated, HeV RNA was not detected in this species.
The absence of any association between pregnancy or lactation and RNA detection, antibody status or lnMFI values, respectively, differs from previous serology-only studies. Plowright et al. [Reference Plowright21] detected significantly higher antibody prevalence in late pregnant/early lactation female little red flying foxes (P. scapulatus) compared with males or non-reproductive females. Breed et al. [Reference Breed22] detected higher antibody prevalence in pregnant and lactating spectacled flying foxes (P. conspicillatus), with titres significantly elevated in pregnant animals. Nonetheless, we did find two notable (and possibly linked) significant associations that suggest the role of female bats in the ecology of HeV infection warrants further focus; namely seropositive females had higher lnMFI values than seropositive males, and viral RNA was more likely to be detected in serum in females than males (Table 1). The latter finding has also been reported in experimentally infected P. alecto [Reference Halpin8]. While our study had large sample sizes overall, high pregnancy rates in adult female bats mean that sample sizes for non-pregnant/non-lactating controls at each time point are limited, constraining the robust interpretation of the effect of sex, pregnancy and lactation from broader seasonal drivers. Specific studies to investigate the mechanisms for such interactions are warranted.
P. alecto had a higher mean antibody prevalence and lnMFI values than did P. poliocephalus. While not unexpected given increasing evidence that the former is a primary reservoir host, the detection of anti-HeV antibodies in over 50% of P. poliocephalus is intriguing, in that it suggests that these individuals have been sufficiently exposed to HeV to mount a humoral response. Yet, in a colony in which HeV is circulating, evidenced by RNA detection in multiple P. alecto, and with nearly half the P. poliocephalus cohort evidently susceptible as indicated by their antibody-negative status, viral RNA was not detected in any P. poliocephalus over the entire study period. Given the epidemiologically ‘adequate’ contact between individuals of the two species when co-roosting, and daily contact with potentially infectious urine [Reference Edson12], plus the substantial sample size, it is highly improbable that the zero detection in P. poliocephalus is a chance event – the upper 95% confidence interval for the population infection rate is 0.5%. The same rationale could be applied to the lack of RNA detection in P. scapulatus, though the argument in this species is somewhat constrained by the smaller sample size, and the uncertain validity of the serology in this species. Targeted studies are required to address variable host species’ responses to henipavirus infection and the various hypotheses put forth below. A plausible explanation, at least in P. poliocephalus, is that the nature of exposure or infection may be adequate to provoke an innate immune response and antibody production, so that the virus is cleared without systemic viral replication and excretion; for example, localised infection and replication of mucous membranes. This scenario is consistent with arguments proposed by Dups et al. [Reference Dups52] and Goldspink et al. [Reference Goldspink11] to explain the aspects of HeV infection dynamics. An alternative interpretation is that the antibody response detected in P. poliocephalus reflects assay cross-reactivity with a closely related henipavirus [Reference Wang53]. This interpretation could explain the lower lnMFI values (reflecting lower binding specificity) observed in P. poliocephalus and the lack of HeV RNA detection even in low BCS P. poliocephalus as discussed above. However, lower lnMFI values in P. poliocephalus might also plausibly reflect a less efficient humoral response associated with non-systemic infection.
The reason for a lack of a clear bimodal distribution of P. scapulatus lnMFI values is unknown, however could suggest cross-reactivity within the serological assay with an unknown Hendra-like virus, a diverse exposure history to HeV within the population, or less plausibly reflect the smaller sample size for this species in this study. Cross-reactivity is a plausible explanation for the detection of anti-HeV antibodies in P. scapulatus given the recent detection of novel henipaviruses in this species [Reference Wang53] and the repeated lack of detection of HeV RNA using a qRT-PCR specific for HeV. Future studies aimed at isolating henipaviruses from P. scapulatus populations are required to clarify this.
Our study is novel in its concurrent molecular and serologic screening of a sample of 1906 bats for evidence of HeV infection, and the longitudinal design has allowed us to explore and explain within-year associations at both event and individual levels with confidence. However, the lack of replicates over multiple years means that inter-annual generalisations should be made with care.
Conclusion
The study has yielded significant new insights into HeV infection and immune dynamics in flying-foxes, as well as provided additional support for a number of previous hypotheses. In summary, our findings support immunologically naïve sub-adult P. alecto playing an important role in maintaining HeV infection at a population level. Contrary to previous studies, we found no association between HeV infection and pregnancy or lactation, and therefore no support for reproductive stress as a driver for infection or recrudescing infection associated with pregnancy. The paradoxical serologic and molecular findings in P. poliocephalus suggest that HeV exposure in this species may not routinely result in systemic infection and virus excretion, or alternatively, that the serologic findings reflect assay cross-reactivity with another henipavirus. Finally, we identify an association between BCS and infection that plausibly could support a role for immune system competence in HeV infection in flying-foxes.
Supplementary material
The supplementary material for this article can be found at https://doi.org/10.1017/S0950268819001237
Author ORCIDs
H. E. Field, 0000-0002-4671-7002
Acknowledgements
We acknowledge and thank the following people and institutions: Biosecurity Queensland officers Craig Smith for project coordination and support, and Paul Willett and Perry Jones at the Boonah office for logistical support; Gary Crameri and colleagues (CSIRO Australian Animal Health Laboratory, Geelong, Australia), Justin Welbergen and colleagues (University of Western Sydney, Australia), Adam McKeown (CSIRO Sustainable Ecosystems, Atherton, Australia) for periodic field assistance; TJ McKinley (University of Exeter, United Kingdom) for further development of the Bayesian mixture model; Batavia Flying Fox Sanctuary (http://www.longgrasssystems.com.au/batavia.html) for age growth data which assisted early analyses; and David Jordan (New South Wales Department of Primary Industries) and Janine Barrett (Queensland Department of Agriculture and Fisheries) for helpful comments on the manuscript.
Financial support
Core funding was provided by the Australian, Queensland and News South Wales governments under the National Hendra virus Research Program; AJP was funded by a Queensland Government Accelerate Post-Doctoral Research Fellowship grant; HEF was supported in part by the United States Agency for International Development (USAID) Emerging Pandemic Threats PREDICT (Cooperative Agreement No. AID-OAA-A-14-00102) and the National Institute of Allergy And Infectious Diseases of the National Institutes of Health (Award No. R01AI110964).







 Open access
Open access