



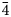
Introduction
Tetrahedrite-group minerals form a complex isotypic series characterised by several homo- and heterovalent substitutions, involving both cations and anions. The general formula of these phases can be written as M (2)A6M (1)(B4C2)X (3)D4S(1)Y12S(2)Z, where A = Cu+, Ag+, □ (vacancy); B = Cu+ and Ag+; C = Zn2+, Fe2+, Hg2+, Cd2+, Ni2+, Mn2+, Cu2+, Cu+, In3+ and Fe3+; D = Sb3+, As3+, Bi3+ and Te4+; Y = S2– and Se2–; and Z = S2–, Se2– and □ (Biagioni et al., Reference Biagioni, George, Cook, Makovicky, Moëlo, Pasero, Sejkora, Stanley, Welch and Bosi2020a). Following this revision of the nomenclature of tetrahedrite-group minerals, a renewed interest in these common sulfosalts, occurring in different kinds of hydrothermal ore deposits, has allowed the identification of several new mineral species and the collection of high-quality crystal chemical data. Usually, members of the tetrahedrite group are characterised by the presence of S as the dominant anion at both the S(1) and S(2) sites. Only three series, i.e. the hakite, ústalečite, and the potential giraudite series, show S replaced by Se, with the aggregate site S(1) + S(2) showing Se > S; along them, another selenide is represented by the unassigned mineral pošepnýite.
Selenium belongs to Group VIA of the Periodic Table along with O, S, Te and Po and is a chalcogen. The chemistry of Se is similar to that of S and, similar to this latter element, it can occur in different oxidation states, i.e. –2, 0, +4 and +6, in some cases forming (Se2)2– groups (e.g. in ferroselite and dzharkenite, dimorphs of the compound FeSe2, isotypic with marcasite and pyrite, respectively). Indeed, the similarity in oxidation states and ionic radii between Se and S favour their reciprocal replacement in several chemical compounds. However, the abundance of Se in the continental crust (120 ng/g – Wedepohl, Reference Wedepohl1995) is much lower than that of S (697 μg/g – Wedepohl, Reference Wedepohl1995) and consequently selenides are rarer than sulfides, notwithstanding a relatively similar diversity index, as defined by Christy (Reference Christy2015).
The first Se-dominant member of the tetrahedrite group, hakite, was described as a new mineral species by Johan and Kvaček (Reference Johan and Kvaček1971) from Předbořice, Central Bohemia region (Czech Republic). The material studied showed variable Se:S atomic ratios, and S-bearing as well as S-free compositions were given, i.e. Cu6(Cu4.07Hg1.83)Σ5.90(Sb3.06As1.03)Σ4.09(Se10.36S2.63)Σ12.99 and Cu6(Cu4.21Hg1.74)Σ5.95(Sb3.83As0.23)Σ4.06Se11.89. These two compositions correspond to unit-cell parameters a = 10.83(1) and 10.88(1) Å, respectively. Johan and Kvaček (Reference Johan and Kvaček1971) proposed the existence of a solid solution between hakite and tetrahedrite. From the same locality, Brodin et al. (Reference Brodin, Osipov, Kachalovskaya, Kozlova and Nazarenko1979) reported the occurrence of Ag-bearing hakite; however, the recalculation of its chemical formula shows that Cu is still the dominant cation at the M(2) site. Spiridonov et al. (Reference Spiridonov, Kachalovskaya and Chvileva1986) described hakite with high Tl (up to 2.56 wt.%) and Ge (up to 1.27 wt.%) contents. Förster et al. (Reference Förster, Rhede and Tischendorf2002) described a complete substitution series between hakite and Hg-rich giraudite from the Niederschlema-Alberoda uranium deposit, Erzgebirge, Germany, whereas Förster and Rhede (Reference Förster and Rhede2004) described the substitutional series between giraudite and tennantite from the same German locality. Hakite was also described from the Zadní Chodov (Scharmová and Scharm, Reference Scharmová and Scharm1995), Černý Důl (Kopecký et al., Reference Kopecký, Pauliš and Škoda2010), Zálesí (Sejkora et al., Reference Sejkora, Macek, Škácha, Pauliš, Plášil and Toegel2014) and Ústaleč deposits, Czech Republic (Sejkora et al., Reference Sejkora, Mauro and Biagioni2023a), Skrikerum, Sweden (Adelmann and Förster, Reference Adelmann and Förster2022) and Tuminico, Sierra de Cacho, Argentina (Paar et al., Reference Paar, Topa, Roberts, Criddle, Amann and Sureda2002). For all the above occurrences, the dominant Me2+ element is Hg, so they correspond to hakite-(Hg) defined by Biagioni et al. (Reference Biagioni, George, Cook, Makovicky, Moëlo, Pasero, Sejkora, Stanley, Welch and Bosi2020a).
Mercury-free hakite (with some Zn contents) was first described from the Bukov deposit, western Moravia, Czech Republic by Kvaček (Reference Kvaček1979); unfortunately, no chemical data or detailed description were published. The not-yet approved ‘hakite-(Cu)’ was mentioned with no chemical data from the Ústaleč deposit, Czech Republic, by Sejkora et al. (Reference Sejkora, Plášil and Makovicky2022a, Reference Sejkora, Biagioni, Škácha, Musetti and Mauro2024).
Cadmium-rich hakite with very close contents of Hg, Cd and Cu2+ was reported by Sejkora et al. (Reference Sejkora, Macek, Škácha, Pauliš, Plášil and Toegel2014) from the deposit Zálesí, Czech Republic. Škácha et al. (Reference Škácha, Palatinus, Sejkora, Plášil, Macek and Goliáš2016) described hakite from Příbram, Central Bohemia (Czech Republic), and stressed the occurrence of different compositions characterised by the dominance of Hg2+, Zn2+, or Cd2+. They indicated these different compositions as ‘Hg-hakite’, ‘Zn-hakite’ and ‘Cd-hakite’, ideally Cu6(Cu4Hg2)Sb4Se13, Cu6(Cu4Zn2)Sb4Se13 and Cu6(Cu4Cd2)Sb4Se13, respectively. The crystal structure of ‘Hg-hakite’ was solved through electron diffraction tomography, confirming the isotypic relations with tetrahedrite and the occurrence of Hg2+ at the M(1) site. Later, Škácha et al. (Reference Škácha, Sejkora and Plášil2017b) also described, in addition to Hg-, Cd-, Zn-members, Fe- and Cu-dominant hakite samples from the same occurrence on the basis of electron microprobe data. Further chemical and crystallographic investigations confirmed such a preliminary identification, allowing the proposal of the three new mineral species hakite-(Cd), hakite-(Fe) and hakite-(Zn) that, along with hakite-(Hg), form the hakite series.
These new minerals and their names were approved by the Commission on New Minerals, Nomenclature and Classification of the International Mineralogical Association (IMA2022–090, 2022–082 and 2022–083, Newsletter 70, Miyawaki et al., Reference Miyawaki, Hatert, Pasero and Mills2023). They are named after their chemical composition in agreement with the nomenclature of the tetrahedrite group (Biagioni et al., Reference Biagioni, George, Cook, Makovicky, Moëlo, Pasero, Sejkora, Stanley, Welch and Bosi2020a). Their mineral symbols, in accord with Warr (Reference Warr2021), are Hak-Cd, Hak-Fe and Hak-Zn. The holotype materials are deposited in the mineralogical collection of the Department of Mineralogy and Petrology of the National Museum, Prague, Czech Republic [polished sections under catalogue numbers P1P 30/2022 for hakite-(Cd) and P1P 11/2016 for both hakite-(Fe) and hakite-(Zn)] and in the mineralogical collection of the Museo di Storia Naturale of the Università di Pisa, Via Roma 79, Calci (PI), Italy [crystals used for the single-crystal X-ray diffraction study under catalogue numbers 20022, 20019 and 20021 for hakite-(Cd), hakite-(Fe) and hakite-(Zn), respectively].
In this paper the description of the new members of the hakite series and details of their crystal structures are reported, along with a discussion on some crystal-chemical and nomenclature issues.
Occurrence and mineral description
Occurrence
Three new members of the hakite series were found at the mine dump of the shaft No. 16 – Háje near Příbram which mined the Bytíz ore deposit of the uranium and base-metal Příbram ore district, Central Bohemia, Czech Republic (GPS coordinates: 49°40'33.894“N 14°3'29.922”E).
The Příbram ore area is famous for its deposits of uranium as well as base-metal ores. There are two large ore districts in the Příbram ore area: the eastward Příbram uranium and base-metal district and the westward lying Pb–Ag Březové Hory ore district (Ettler et al., Reference Ettler, Sejkora, Drahota, Litochleb, Pauliš, Zeman, Novák and Pašava2010). The first represents the most considerable accumulation of vein-type hydrothermal U ores in the Czech Republic (with the production of 48.432 t of pure U metal) and is comparable to world-class deposits of this type. The hydrothermal U mineralisation of Late Variscan age is related to a 1–2 km wide and almost 25 km long zone formed by a strongly tectonised series of Upper Proterozoic rocks along with the contact with granitoids of the Early Carboniferous Central Bohemian Plutonic Complex (Litochleb et al., Reference Litochleb, Černý, Litochlebová, Sejkora and Šreinová2003). Beside economic importance, the Příbram ore area is also marked by an incredible mineralogical diversity, the full list of minerals recorded there (more than 300 species) is given at http://www.mindat.org/loc-779.html.
In the Příbram uranium and base-metal district, there are four main mineralisation stages: (1) siderite-sulfidic; (2) calcite; (3) calcite–uraninite; and (4) calcite-sulfidic. Selenide mineralisation occurs in close association with uraninite of the calcite–uraninite mineralisation, but selenides are always younger than uraninite. Uranium ore is represented by uraninite, coffinite and U-bearing anthraxolite (pyrobitumen). The age of the uranium mineralisation, obtained by U–Pb radiometric age determination of uraninite, is Lower Permian, i.e. 275 ± 4 and 278 ± 4 Ma (Anderson, Reference Anderson1987). The Se–U mineralisation is of low-temperature hydrothermal origin and is associated with calcite veins, tens of centimetres to several metres thick. The observed selenide assemblage is extraordinarily rich, with the total number of selenide species exceeding 25 (Litochleb et al., Reference Litochleb, Sejkora and Šrein2004; Škácha and Sejkora, Reference Škácha and Sejkora2007; Škácha et al., Reference Škácha, Sejkora, Litochleb and Hofman2009, Reference Škácha, Buixaderas, Plášil, Sejkora, Goliáš and Vlček2014, Reference Škácha, Plášil, Sejkora and Goliáš2015, Reference Škácha, Palatinus, Sejkora, Plášil, Macek and Goliáš2016, Reference Škácha, Sejkora and Plášil2017b; Sejkora et al., Reference Sejkora, Škácha, Laufek and Plášil2017, Reference Sejkora, Buixaderas, Škácha and Plášil2018) and including the recently approved new species příbramite (Škácha et al., Reference Škácha, Sejkora and Plášil2017a), bytízite (Škácha et al., Reference Škácha, Sejkora and Plášil2018), and pošepnýite (Škácha et al., Reference Škácha, Sejkora, Plášil and Makovicky2020), along with the three new members of the hakite series (this paper). More details on the selenide mineral associations in the Příbram uranium and base-metal district and conditions of their origin are given in Škácha et al. (Reference Škácha, Sejkora and Plášil2017b).
New members of the hakite series were identified in calcite-uraninite gangue. Hakite-(Cd) occurs in close association with clausthalite, cadmoselite and hakite-(Hg). Hakite-(Fe) and hakite-(Zn) were characterised from the selenide-rich assemblage in association with berzelianite, bukovite, bytízite, crookesite, chaméanite, eskebornite, příbramite, the not yet approved ‘giraudite-(Hg)’ and ‘giraudite-(Cu)’, hakite-(Hg), umangite and a new Cu–As selenide. Their crystallisation is related to the circulation of hydrothermal fluids with temperature near 100°C in the neutral-to-weakly-alkaline environment with high $f_{\rm O_2}$ and a high $f_{\rm Se_2}$
and a high $f_{\rm Se_2}$ /$f_{\rm S_2}$
/$f_{\rm S_2}$ ratio (Škácha et al., Reference Škácha, Sejkora and Plášil2017b).
ratio (Škácha et al., Reference Škácha, Sejkora and Plášil2017b).
Physical and optical properties
Hakite-(Cd) occurs as anhedral grains and veinlets up to 100 μm in size (Fig. 1a,b). It is locally replaced by lath-shaped crystals of cadmoselite, the replacement starting from the centre of aggregates.

Figure 1. Back-scattered electron (BSE) (a,c,e) and reflected light (b,d,f) images of the studied samples of hakite-(Cd) (a,b), hakite-(Fe) (c,d) and hakite-(Zn) (e,f). The grains used for single-crystal X-ray diffraction study were extracted from the red boxes. Holotype samples (see text).
Hakite-(Fe) occurs as anhedral grains, up to 80 × 300 μm in size (Fig. 1c,d), in association with chalcopyrite, ‘hakite-(Cu)’, chaméanite, ‘giraudite-(Cu)’, permingeatite, tetrahedrite-(Fe), bukovite, bytízite and příbramite.
Finally, hakite-(Zn) was observed as irregular aggregates up to several mm in size. It also forms, along with hakite-(Hg) and tetrahedrite-(Zn), domains within zoned idiomorphic crystals up to 100 μm in size (Škácha et al., Reference Škácha, Palatinus, Sejkora, Plášil, Macek and Goliáš2016). In the sample used for single-crystal X-ray diffraction study, hakite-(Zn) occurs as anhedral grains up to 60 × 200 μm in size (Fig. 1e,f).
The three new members of the hakite series are black in colour and opaque in transmitted light; they have a metallic lustre. The Mohs hardness is close to 3½–4, in agreement with other members of the tetrahedrite group. They are brittle, with an indistinct cleavage and a conchoidal fracture. Calculated density is 6.019 (Hak-Cd), 6.011 (Hak-Fe) and 6.081 g.cm–3 (Hak-Zn). In reflected light, they are isotropic, pale grey with bluish (Hak-Cd) or brownish (Hak-Fe and Hak-Zn) shades. Internal reflections were not observed. Reflectance spectra of these new species were measured in air with a TIDAS MSP400 spectrophotometer attached to a Leica microscope (100× objective) using a WTiC (Zeiss No. 370) standard, with a square sample measurement field of ca. 5 × 5 μm. The results for the 400–700 nm range are given in Table 1 and plotted in Fig. 2 in comparison with published data for hakite-(Hg).
Table 1. Reflectance values (%) for the new members of the hakite series.*

* The reference wavelengths required by the Commission on Ore Mineralogy (COM) are given in bold.

Figure 2. Reflectivity curves for members of the hakite series; hakite-(Cd), hakite-(Fe) and hakite-(Zn) (this paper); hakite-(Hg)1 – Picot and Johan (Reference Picot and Johan1982), hakite-(Hg)2 – Criddle and Stanley (Reference Criddle and Stanley1993 – page 218), hakite-(Hg)3 Criddle and Stanley (Reference Criddle and Stanley1993 – page 219).
Chemical composition
Chemical analyses were performed using a Cameca SX100 electron microprobe (National Museum, Prague) operating in wavelength-dispersive mode (25 kV, 20 nA and 1 μm wide beam). The following standards and X-ray lines were used to minimise line overlaps: Ag (AgLα); Au (AuMα); Bi (BiMβ); CdTe (CdLα); Co (CoKα); chalcopyrite (CuKα and SKα); pyrite (FeKα); HgTe (HgMα); NiAs (NiKα, AsLβ); PbS (PbMα); PbSe (SeLα); PbTe (TeLα); Sb2S3 (SbLα); Tl(Br,I) (TlLα) and ZnS (ZnKα). Peak counting times were 20 s for all elements and one half of the peak time for each background. Gold, Bi, Co, Ni, Pb, Te and Tl were found to be below the detection limits (0.02–0.10 wt.%). Raw intensities were converted to the concentrations of elements using the automatic ‘PAP’ (Pouchou and Pichoir, Reference Pouchou, Pichoir and Armstrong1985) matrix-correction procedure.
Experimental data for the three new members of the hakite series are given in Table 2. Their empirical chemical formulae were recalculated according to ΣMe = 16 atoms per formula unit (apfu), assuming that no vacancies occur at the M(2), M(1) and X(3) sites.
Table 2. Chemical data (wt.%) for the new members of the hakite series.
(σ) = estimated standard deviation; n = number of spot analyses.
The empirical formula of hakite-(Cd) is Cu9.71(8)Ag0.24(7)Cd1.51(2)Hg0.43(2)Zn0.03(1)(Sb3.94(7)As0.13(7))Σ4.07Se11.35(24)S1.57(24), corresponding to the end-member formula Cu6(Cu4Cd2)Sb4Se13, which requires (in wt.%) Cu 26.77, Cd 9.47, Sb 20.52, Se 43.24, total 100.00.
Hakite-(Fe) and hakite-(Zn) have empirical chemical formulae Cu10.11(7)Ag0.18(2)Fe0.81(2)Zn0.50(5)Hg0.26(2)(Sb3.72(13)As0.41(10))Σ4.13Se12.65(12)S0.12(2) and Cu10.03(4)Ag0.24(2)Zn0.61(2)Fe0.53(2)Hg0.45(4)(Sb3.55(8)As0.60(7))Σ4.15Se12.82(6)S0.08(4), respectively, corresponding to the end-member formulae Cu6(Cu4Fe2)Sb4Se13 and Cu6(Cu4Zn2)Sb4Se13, respectively. These formulae require (in wt.%) Cu 28.11, Fe 4.94, Sb 21.54, Se 45.41, total 100.00, for hakite-(Fe), and Cu 27.88, Zn 5.74, Sb 21.36, Se 45.03, total 100.00, for hakite-(Zn).
X-ray diffraction data
Grains of the new members of the hakite series were extracted from the polished sections previously studied through electron microprobe analysis (Fig. 1) and mounted on a carbon fibre to be examined with a Bruker D8 Venture single-crystal X-ray diffractometer equipped with an air-cooled Photon III area detector and microfocus MoKα radiation (Centro per l'Integrazione della Strumentazione Scientifica dell'Università di Pisa, University of Pisa). The detector-to-crystal distance was set to 42 mm for hakite-(Cd) and 38 mm for both hakite-(Fe) and hakite-(Zn) and data were collected using φ scan modes in 0.5° slices. Data were integrated using a narrow-frame algorithm and they were corrected for Lorentz-polarisation, absorption, and background using the software package Apex 4 (Bruker AXS, 2022). The crystal structures of the studied species were refined using Shelxl-2018 (Sheldrick, Reference Sheldrick2015), starting from the atomic coordinates of Johnson and Burnham (Reference Johnson and Burnham1985). The occurrence of a racemic twin was modelled and the crystal structure of all the three hakite-series members needed to be inverted. Neutral scattering factors were taken from the International Tables for Crystallography (Wilson, Reference Wilson1992). Crystal structure refinements are described below, whereas further details on data collection and refinement are given in Table 3. Fractional atom coordinates and equivalent isotropic parameters are reported in Table 4, whereas Table 5 shows selected bond distances. Table 6 compares the mean atomic numbers observed during the crystal structure refinement with those calculated on the basis of electron microprobe data. Finally, weighted bond-valence sums, calculated using the bond-parameters of Brese and O'Keeffe (Reference Brese and O'Keeffe1991) and the site populations shown in Table 6, are given in Table 7. For all the three new hakites, crystallographic information files (cifs) have been deposited with the Principal Editor of Mineralogical Magazine and are available as Supplementary material (see below).
Table 3. Summary of data collection and refinement parameters for the new members of the hakite series.

1 w = 1/[σ2(F o2)+(0.0224P)2+6.0568P] for hakite-(Cd); w = 1/[σ2(F o2)+(0.0205P)2+9.5395P] for hakite-(Fe); w = 1/[σ2(F o2)+(0.0232P)2+17.8803P] for hakite-(Zn).
2 Flack (Reference Flack1983).
Table 4. Site, Wyckoff positions, site occupancy (s.o.f.), fractional atom coordinates, equivalent isotropic displacement parameters (Å2) for the new members of the hakite series.

Table 5. Selected bond distances (in Å) for new members of the hakite series.

Table 6. Refined mean atomic numbers (MAN – in electrons) and proposed site occupancies for cation and anion sites in the new members of the hakite series.
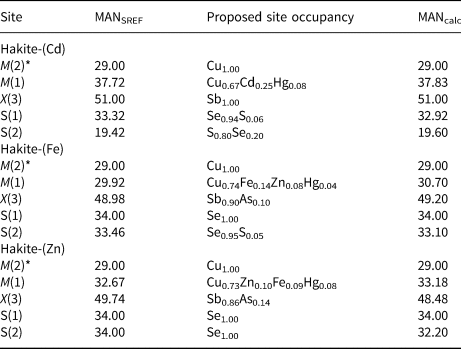
* hakite-(Cd) M(2) = M(2a) + 2M(2b); hakite-(Fe) M(2) = M(2a) + 2M(2b); hakite (Zn) M(2) = M(2a) + 2M(2b)
Table 7. Weighted bond-valence sums (in valence units) in the new members of the hakite series

Powder X-ray diffraction data for new members of the hakite series could not be collected, due to the paucity of available material. Consequently, powder X-ray diffraction data, given in Table 8, were calculated using the software PowderCell 2.3 (Kraus and Nolze, Reference Kraus and Nolze1996) on the basis of the structural models given in Tables 3 and 4.
Table 8. Calculated powder X-ray diffraction data for the new members of the hakite series.*

* Intensity and d hkl (in Å) were calculated using the software PowderCell2.3 (Kraus and Nolze, Reference Kraus and Nolze1996) on the basis of the structural data given in Tables 3 and 4. Only reflections with I rel. ≥ 1 are listed. The five strongest reflections are given in bold.
Hakite-(Cd)
Intensity data were collected using a short prismatic fragment, 60 × 60 × 40 μm in size. A total of 544 frames was collected with an exposure time of 10 s per frame. Unit-cell parameters, refined on the basis of the XYZ centroids of 1356 reflections above 20 σI with 7.49 < 2θ < 59.34°, are a = 10.8860(6) Å and V = 1290.0(2) Å3. The following scattering curves were used initially: Cu at M(2), Cu vs. Cd at M(1), Sb vs. As at X(3), Se vs. S at S(1) and S(2). An isotropic model converged to R 1 = 0.0801, thus confirming the correctness of the structural model. The site occupancy refined at the X(3) site indicated that it is a virtually pure Sb position, whereas S(1) and S(2) were found to be Se- and S-dominant sites, respectively. The highest residuals were located around the M(2) site, suggesting its splitting. The addition of the split position M(2b) lowered the R 1 value to 0.0585. The anisotropic structural model converged to R 1 = 0.0230 for 358 reflections with F o > 4σ(F o) and 24 refined parameters.
Hakite-(Fe)
Intensity data were collected using a fragment reaching 85 × 60 × 35 μm in size. A total of 552 frames was collected with an exposure time of 5 s per frame. Unit-cell parameters, refined on the basis of the XYZ centroids of 2251 reflections above 20 σI with 7.55 < 2θ < 64.92°, are a = 10.7983(4) Å and V = 1259.12(14) Å3. The following scattering curves were used during the first stages of refinement: Cu at M(2), Sb vs. As at X(3), Se vs. S at S(1) and S(2). Electron microprobe data suggests that the M(1) site may have a site occupancy close to (Cu0.74Fe0.14Zn0.08Hg0.04). Owing to the similarity of the scattering factors of Cu (Z = 29), Fe (Z = 26), and Zn (Z = 30), the site occupancy at the M(1) site was refined using the scattering factors of Cu vs. Hg. An isotropic model converged to R 1 = 0.0947, with a high residual of ~ 8 e/Å3 close to M(2), suggesting its splitting. Moreover, the refinement of the site occupancies at the S(1) and S(2) sites indicated that the former corresponds to a virtually pure Se position, whereas the latter has a minor S content, in agreement with electron microprobe data indicating very low amounts of S. After the splitting of the M(2) site, the refinement converged to R 1 = 0.0440. The anisotropic structural model converged to R 1 = 0.0254 for 431 reflections with F o > 4σ(F o) and 24 refined parameters.
Hakite-(Zn)
Intensity data were collected using a short prismatic fragment, 70 × 60 × 60 μm in size. A total of 560 frames was collected with an exposure time of 10 s per frame. Unit-cell parameters, refined on the basis of the XYZ centroids of 1229 reflections above 20 σI with 7.54 < 2θ < 59.79°, are a = 10.8116(14) Å and V = 1263.8(5) Å3. The following scattering curves were used during the first stages of refinement: Cu at M(2); Sb vs. As at X(3); and Se vs. S at S(1) and S(2). Electron microprobe data suggests that the M(1) site may have a site occupancy close to (Cu0.73Zn0.10Fe0.09Hg0.08). As for hakite-(Fe), the site occupancy at the M(1) site was refined using the scattering factors of Cu vs. Hg. An isotropic model converged to R 1 = 0.0925, with a high residual of ~ 9 e–/Å3 close to M(2), suggesting its splitting. The refinement of the site occupancies at the S(1) and S(2) positions indicated that both are virtually pure Se positions, in agreement with chemical data showing a very low content of S. After the splitting of the M(2) site, the refinement converged to R 1 = 0.0464. The anisotropic structural model converged to R 1 = 0.0302 for 353 reflections with F o > 4σ(F o) and 23 refined parameters.
Results and discussion
Crystal structures of new members of the hakite series
The crystal structures of the new minerals belonging to the hakite series are isotypic with those of the other members of the tetrahedrite group (Biagioni et al., Reference Biagioni, George, Cook, Makovicky, Moëlo, Pasero, Sejkora, Stanley, Welch and Bosi2020a), i.e. they show a three-dimensional framework formed by corner-sharing M(1)-centred tetrahedra with cages hosting S(2)-centred M(2)6-octahedra, encircled by four X(3)S(1)3 trigonal pyramids. As observed in other tetrahedrite-group isotypes (e.g. Andreasen et al., Reference Andreasen, Makovicky, Lebech and Karup-Møller2008; Welch et al., Reference Welch, Stanley, Spratt and Mills2018), the M(2) site is split into two sub-positions, namely M(2a) and M(2b). The former has a triangular planar coordination, whereas the latter has a flat trigonal pyramidal coordination.
Cation sites
Three cation sites occur in the crystal structure of tetrahedrite isotypes, namely the M(2), M(1) and X(3) sites.
In the crystal structures of the new members of the hakite series, the electron density associated with the M(2) site can be split into two sub-positions, as stated above. These positions cannot be occupied simultaneously, owing to short M(2a)–M(2b) distances, i.e. 0.585(12), 0.679(7) and 0.705(11) Å in hakite-(Cd), hakite-(Fe) and hakite-(Zn), respectively. Moreover, the distance of two neighbouring M(2b) positions is 1.17(2), 1.35(214) and 1.40(2) Å for the Cd-, Fe-, and Zn-members of the hakite series. The average cation–anion bond distances are shorter for the M(2a) positions than for the M(2b) sites. Indeed, the <M(2a)–φ> distances (where φ = Se and S) are 2.334, 2.345 and 2.344 Å for hakite-(Cd), hakite-(Fe) and hakite-(Zn), respectively, whereas the corresponding <M(2b)–φ> distances are 2.402, 2.432 and 2.437 Å. The shorter M(2b)–φ distance observed for hakite-(Cd) is probably due to its higher S content, with S being smaller than Se.
In the samples studied, both M(2a) and M(2b) positions are mainly occupied by Cu, as the Ag content is negligible (less than 0.04 Ag apfu). Usually, in tetrahedrite-group minerals, the mean atomic number refined at the M(2a) site is higher than that at M(2b), indicating that the cations are hosted preferentially at the former position (e.g. in tennantite-(Cu) – Biagioni et al., Reference Biagioni, Sejkora, Moëlo, Marcoux, Mauro and Dolníček2022a; tetrahedrite-(Cd) – Sejkora et al., Reference Sejkora, Biagioni, Škácha, Musetti, Kasatkin and Nestola2023b), whereas in some refinement the splitting was not observed nor modelled (e.g. in kenoargentotetrahedrite-(Fe) – Shu et al., Reference Shu, Shen, Lu and Gu2022; stibiogoldfieldite – Biagioni et al., Reference Biagioni, Sejkora, Musetti, Makovicky, Pagano, Pasero and Dolníček2022b; tetrahedrite-(Hg) – Biagioni et al., Reference Biagioni, Sejkora, Musetti, Velebil and Pasero2020b). On the contrary, something interesting occurs in the hakite-series minerals. Indeed, taking into account site multiplicity, the M(2) site population of hakite-(Cd) is M (2)[M (2a)Cu3.54M (2b)Cu2.46], whereas in hakite-(Fe) and hakite-(Zn) the site populations are M (2)[M (2a)Cu2.63M (2b)Cu3.37] and M (2)[M (2a)Cu2.82M (2b)Cu3.18], respectively. This suggests that in the two latter phases, Cu has a slight preference for the M(2b) position. It is worth noting what happens in another selenide series belonging to the tetrahedrite group, i.e. the ústalečite series (Sejkora et al., Reference Sejkora, Plášil and Makovicky2022a, Reference Sejkora, Biagioni, Škácha, Musetti and Mauro2024). In arsenoústalečite and stibioústalečite the site population at the M(2) site can be written as M (2)[M (2a)Cu4.55M (2b)Cu1.45] and M (2)[M (2a)Cu4.98M (2b)Cu1.02] (Sejkora et al., Reference Sejkora, Biagioni, Škácha, Musetti and Mauro2024). The preference of Cu for the M(2a) or M(2b) positions seems to be related (at least partially) to the occupancy at the S(2) site. Indeed, whenever S(2) is occupied by S (or dominated by S), Cu prefers M(2a); on the contrary, when S(2) site is a Se-dominant position, M(2b) seems to be preferred. This could be due to the necessity to avoid too short Cu–Se bond distances. Indeed, triangularly coordinated Cu forms bond distances with Se of ca. 2.40 Å (e.g., bytízite – Škácha et al., Reference Škácha, Sejkora and Plášil2018), in agreement with the M(2a)–S(1) and M(2b)–S(1) distances ranging between 2.39 and 2.44 Å. On the contrary, the bond distance M(2a)–S(2) is definitely short, varying between 2.22 and 2.24 Å, whereas the M(2b)–S(2) distances range between 2.33 Å for the S-dominant S(2) site of hakite-(Cd) and 2.41–2.42 Å for the Se-dominant S(2) sites in the Fe- and Zn-isotypes of the hakite series. Bond valence sums at the [M(2a) + 2M(2b)] sites are 1.06, 1.12 and 1.10 valence units (v.u.) for hakite-(Cd), hakite-(Fe) and hakite-(Zn), respectively, in agreement with the occurrence of monovalent cations at M(2) (Table 7).
The tetrahedrally coordinated M(1) site, in the three new members of the hakite series, shows different features related to the variable chemistry. This site hosts several transition elements, i.e. Cu, Cd, Fe, Hg and Zn. It is known that Cu and Fe can assume, in tetrahedrite-group minerals, different formal oxidation states, i.e. Cu+, Cu2+, Fe2+ and Fe3+. However, according to Karup-Møller and Makovicky (Reference Karup-Møller and Makovicky1999), in Se-tetrahedrite Fe could not occur as Fe3+ and Cu may be present as Cu+ only. The refined mean atomic numbers (MAN) at the M(1) site in the three minerals studied is in accord with the site occupancies proposed on the basis of electron microprobe data (Table 6). On the contrary, bond-valence sums (Table 7) show a systematic overbonding, with values ranging between 1.68 and 1.80 v.u. This is probably due to the low accuracy of the bond parameters involving Se reported by Brese and O'Keeffe (Reference Brese and O'Keeffe1991) and possibly also to the significant covalency of the metal–selenium bonds. As regards the goodness of the bond parameters, one could observe that their predictive power about the average bond distances is very low. For instance, in hakite-(Cd), one could calculate, on the basis of the site occupancy (Cu0.67Cd0.25Hg0.08), a distance <M(1)–Se> of 2.579 Å, definitely larger than the observed value of 2.492 Å. For this reason, a prediction of the expected average bond distance for the tetrahedrally-coordinated atoms hosted at the M(1) site could be based on experimental distances reported for simple selenides. Still using the case of hakite-(Cd), one could use the following bond distances: Cu–Se 2.421 Å (eskebornite – Delgado et al., Reference Delgado, Diaz de Delgado, Quintero and Woolley1992), Cd–Se 2.634 Å (cadmoselite – Wyckoff, Reference Wyckoff1963), Hg–Se 2.634 Å (tiemannite – Wyckoff, Reference Wyckoff1963). In this case, assuming the site occupancy reported above, an average distance of 2.491 Å can be calculated, close to the observed value of 2.492 Å. Iron and Zn have a smaller atomic radius than Cd, and indeed the <M(1)–Se> distances are shorter, i.e. 2.439 and 2.448Å for hakite-(Fe) and hakite-(Zn), the relatively longer length of the latter species being due to its higher Hg content. Also for these two phases, a prediction of the <M(1)–Se> distances may be based on experimental distances. In addition to those given above, one could use the following values: Fe–Se 2.396 Å (eskebornite – Delgado et al., Reference Delgado, Diaz de Delgado, Quintero and Woolley1992), and Zn–Se 2.454 Å (stilleite – Wyckoff, Reference Wyckoff1963). Using them, and the site occupancies reported in Table 6, the distances of 2.429 and 2.439 Å can be calculated for hakite-(Fe) and hakite-(Zn), respectively, to be compared with the observed values given above.
The X(3) site displays the typical trigonal pyramidal coordination shown by Sb3+ and As3+ in sulfosalts. In the three specimens studied, Sb is the dominant chemical constituent at this site, as shown by both electron microprobe analyses and crystal structure refinements. The Sb/(Sb+As) atomic ratios, as derived from chemical data, are 0.97, 0.90 and 0.86 for hakite-(Cd), hakite-(Fe) and hakite-(Zn), respectively. Such values can be compared with the results of crystal structure refinements, pointing to ratios of 1.00, 0.88 and 0.93. The Sb–(Se,S) distances range between 2.557 and 2.551 Å, the longest distance being observed in the almost Sb pure X(3) site of hakite-(Cd). Bond-valence sums vary between 3.00 and 3.09 v.u., in accord with the presence of trivalent semi-metals (Table 7).
Anion sites
Two anion sites, namely S(1) and S(2), occur in the crystal structure of tetrahedrite-group minerals. The anion hosted at the S(1) position is tetrahedrally coordinated by two M(1), one M(2) [= M(2a) or one of the two mutually exclusive M(2b) positions], and one X(3) sites, whereas the anion at S(2) shows an octahedral coordination, being bonded to six atoms hosted at M(2).
The S(1) site is, in all the studied specimens, a Se-dominant position. In particular, it is probably a Se-pure site in hakite-(Fe) and hakite-(Zn), whereas it shows a minor replacement of Se by S in hakite-(Cd), with a refined Se/(Se+S) atomic ratio of 0.96, corresponding to the site population S(1)(Se11.52S0.48). The bond-valence sums at S(1) range between 2.18 and 2.28 v.u. (Table 7).
The S(2) site is a S-dominant position in hakite-(Cd), having the refined site occupancy (S0.81Se0.19). On the contrary, in the other two members of the hakite series, S(2) is a Se-dominant site, with only a very minor replacement of S in hakite-(Fe), with a refined site occupancy of (Se0.97S0.03). The bond-valence sums at the Se(2) site deserve further discussion. Indeed, this value is physically reasonable at the S(2) site of hakite-(Cd), where this is a S-dominant site, and the bond-valence sum is 2.16 v.u. On the contrary, hakite-(Fe) and hakite-(Zn) show a significant overbonding of the anion atom at the S(2) site, with values of 2.64 v.u. This high value is due to the short M(2)–S(2) distances. Indeed, the splitting of the M(2) into the M(2a) and M(2b) positions, with the latter being preferred in hakite-(Fe) and hakite-(Zn), is not enough to decrease the overbonding. Moreover, as discussed above for the bond-valence sums at the M(1) site, the bond-parameters of Brese and O'Keeffe (Reference Brese and O'Keeffe1991) are probably not very accurate for the Cu–Se bonds. For instance, considering the experimental Cu–Se distance of 2.421 Å observed by Delgado et al. (Reference Delgado, Diaz de Delgado, Quintero and Woolley1992) in eskebornite, one could estimate an ideal R ij of 1.91 Å, to be compared with the tabulated value of 2.02 Å. Using the estimated value of R ij, the bond-valence sums at S(2) in hakite-(Fe) and hakite-(Zn) would be 1.92 and 1.98 v.u., respectively, in agreement with the expected value. Finally, whereas in the crystal structure refinement of hakite-(Cd) there are no significant residuals around S(2), in hakite-(Fe) and hakite-(Zn) such residuals occur (Table 3), and the U eq value at S(2) is higher than that observed in hakite-(Cd). This could indicate a positional disorder of S(2) between the 2a position (at 0, 0, 0) and a 8c position (at x, x, x), with x = 0.0324 and 0.0368 for hakite-(Fe) and hakite-(Zn), respectively. This is similar to that observed by Sejkora et al. (Reference Sejkora, Biagioni, Števko, Raber, Roth and Vrtiška2022b) in argentotetrahedrite-(Zn) and due to the necessity of avoiding too short metal–anion distances.
Finally, it is noteworthy to compare the S/(S+Se) atomic ratios calculated from electron microprobe data and those obtained through crystal structure refinement. In hakite-(Cd), the S-richest specimen among the studied ones, this value is 0.122, to be compared with the refined value, i.e., 0.097. Hakite-(Fe) and hakite-(Zn) are S-poor, with S/(S+Se) atomic ratios of 0.009 and 0.006, respectively. In the former, minor S was refined at the S(2), with a ratio of 0.002, whereas in the latter S was not detected in the crystal structure refinement. This could indicate that, even if S is preferentially partitioned at the S(2) site, some S can be also disordered at S(1), where it could be more difficult to detect low amounts of S owing to its multiplicity. In the pair arsenoústalečite – stibioústalečite, the comparison of S/(S+Se) atomic ratios between chemical and structural data indicated a good agreement for the As-isotype and an underestimation of S in the Sb-isotype (Sejkora et al., Reference Sejkora, Biagioni, Škácha, Musetti and Mauro2024).
Crystal chemistry of members of the hakite series and nomenclature issues
The structural investigation of the three new members of the hakite series led to the following structural formulae, based on both electron microprobe and structural data: hakite-(Cd) = M (2)[M (2a)Cu3.54M (2b)Cu2.46]Σ6.00M (1)(Cu4.02Cd1.50Hg0.48)Σ6.00X (3)Sb4.00S(1)(Se11.28S0.72)Σ12.00S(2)(S0.80Se0.20)Σ1.00; hakite-(Fe) =M (2)[M (2a)Cu2.63M (2b)Cu3.37]Σ6.00M (1)(Cu4.44Fe0.84Zn0.48Hg0.24)Σ6.00X (3)(Sb3.60As0.40)Σ4.00S(1)Se12.00S(2)(Se0.95S0.05)Σ1.00; and hakite-(Zn) = M (2)[M (2a)Cu2.82M (2b)Cu3.18]Σ6.00M (1)(Cu4.38Zn0.60Fe0.54Hg0.48)Σ6.00X (3)(Sb3.44As0.56)Σ4.00S(1)Se12.00S(2)Se1.00. These formulae correspond to the end-member formulae Cu6(Cu4Cd2)Sb4Se12S, Cu6(Cu4Fe2)Sb4Se12Se and Cu6(Cu4Zn2)Sb4Se12Se for the studied samples of hakite-(Cd), hakite-(Fe), and hakite-(Zn), respectively. These end-member formulae do not agree with those given in the formal definition of these three species, where only Se is considered.
In particular, the end-member formula calculated for the type specimen of hakite-(Cd) is reminiscent of those found in the specimens of arsenoústalečite and stibioústalečite, showing the dominance of S at the S(2) site and Se at the S(1) position (Sejkora et al., Reference Sejkora, Biagioni, Škácha, Musetti and Mauro2024). The preference of Se for the S(1) site also agrees with the results of Pohl et al. (Reference Pohl, Ließmann and Okrugin1996).
As shown in Fig. 3, the S/(S+Se) contents in hakite-(Cd), hakite-(Fe), and hakite-(Zn), as well as in hakite-(Hg), are very variable, in some cases approaching the 0.5 value. The observed ranges of S are 0.67–4.53 apfu, 0.06–6.16 apfu, and 0.05–4.42 apfu for the Cd-, Fe-, and Zn-members of the hakite series, respectively (Fig. 3a – our data, Škácha et al., Reference Škácha, Sejkora and Plášil2017b). This means that it is likely to have samples with S(1) and S(2) both occupied by Se and samples where S(1) and S(2) are dominated by Se and S, respectively. This crystal chemical feature opens a nomenclature issue, already discussed in Sejkora et al. (Reference Sejkora, Biagioni, Škácha, Musetti and Mauro2024) for the ústalečite series. Indeed, following Nickel and Grice (Reference Nickel and Grice1998), the compositions S(1)Se12S(2)S and S(1)Se12S(2)Se should correspond to different mineral species because “at least one structural site […]” is “predominantly occupied by a different chemical component than that which occurs in the equivalent site in an existing mineral species”. On the contrary, the current nomenclature of the tetrahedrite group (Biagioni et al., Reference Biagioni, George, Cook, Makovicky, Moëlo, Pasero, Sejkora, Stanley, Welch and Bosi2020a) considers the dominance at the aggregate site S(1) + S(2), in order to avoid a further “proliferation” of mineral species. It is clear that the different partitioning of Se and S between these two sites is not trivial, but it plays an important role in avoiding too short M(2)–S(2) bond distances. However, as discussed above, this is not the only mechanism, and S-free selenides belonging to the hakite series occur in Nature. In fact, the range of SeS–1 substitution in these minerals is probably unlimited, the observed gaps being more related to the amount of samples available for study than to any fields of immiscibility. A similar range of SeS–1 substitution was also observed for hakite-(Hg) (Fig. 3b). The sample studied by Škácha et al. (Reference Škácha, Palatinus, Sejkora, Plášil, Macek and Goliáš2016) has 1.12 S apfu but unfortunately the structural data, collected using electron diffraction tomography, were probably not good enough (R = 24.4%) to identify the actual distribution of S between S(1) and S(2).

Figure 3. Compositional variation (in apfu) of hakite-series minerals: (a) Se vs S for hakite-(Cd); -(Fe) and -(Zn) from Příbram, our data, Škácha et al. (Reference Škácha, Sejkora and Plášil2017b); (b) Se vs S for hakite-(Hg), our data, Škácha et al. (Reference Škácha, Sejkora and Plášil2017b), Johan and Kvaček (Reference Johan and Kvaček1971), Brodin et al. (Reference Brodin, Osipov, Kachalovskaya, Kozlova and Nazarenko1979), Spiridonov et al. (Reference Spiridonov, Kachalovskaya and Chvileva1986), Scharmová and Scharm (Reference Scharmová and Scharm1995), Paar et al. (Reference Paar, Topa, Roberts, Criddle, Amann and Sureda2002), Kopecký et al. (Reference Kopecký, Pauliš and Škoda2010), Sejkora et al. (Reference Sejkora, Macek, Škácha, Pauliš, Plášil and Toegel2014); and (c) As vs. Ag for hakite-(Cd), -(Fe) and -(Zn) from Příbram, our data, Škácha et al. (Reference Škácha, Sejkora and Plášil2017b).
For the sake of simplicity, S(1) and S(2) sites should be considered as an aggregate site for nomenclature purposes, avoiding the creation of different isotypes of these minerals based on different S/Se ratios. On the contrary, species with S > Se should be classified as tetrahedrite (tennantite/goldfieldite), and those with Se > S could be described as members of the hakite (giraudite/ústalečite) series. However, owing to the interesting crystal chemical behaviour of S and Se, an adjectival modifier may be used to highlight high S contents, e.g., S-bearing hakite-(Cd).
In the hakite series, the most important substitution seems to occur at the M(1) and S(2) sites. However, M(2) and X(3) sites could be involved in some other substitution, typical of tetrahedrite-group minerals. For instance, Cu could be replaced by Ag at the M(2) site even if, in the three studied specimens, the Ag content is negligible and, as shown in Figure 3c, it is usually below 1 apfu. Similarly, the X(3) site may show the partial replacement of Sb by As (e.g., Förster et al., Reference Förster, Rhede and Tischendorf2002). However, also in this case, chemical data available for the studied material indicate only a limited replacement, with As contents lower than 1 apfu (Fig. 3c).
Conclusions
The discovery of hakite-(Cd), hakite-(Fe) and hakite-(Zn) and the refinement of their crystal structures improve our knowledge of selenide members of the tetrahedrite group based on high-quality electron microprobe analyses and single-crystal X-ray diffraction data. So far, the only available structural data were those collected by Škácha et al. (Reference Škácha, Palatinus, Sejkora, Plášil, Macek and Goliáš2016) for hakite-(Hg) based on electron diffraction data, giving a very high R value, and therefore not enabling us to decipher the subtle structural features of this complex selenosalt.
The investigation of the three new hakite-series minerals allows us to understand the different mechanisms favouring the avoiding of too short Me–Se distance at the S(2) site, with both the preferential partitioning of S (where available) at this site, as reported by Sejkora et al. (Reference Sejkora, Biagioni, Škácha, Musetti and Mauro2024) in the ústalečite series, or the splitting of the M(2) site, with an increase in population of the 24g position showing longer Me–Se distances and a possible disordering of Se at a 8c position. Probably, the investigation of further specimens of selenide members of the tetrahedrite group, including hakite-(Hg) for which high-quality data are still lacking, would allow us to refine our understanding of the crystal chemical mechanisms controlling the structural plasticity of this important group of sulfo- and selenosalts.
Acknowledgements
The helpful comments of reviewers Peter Leverett and Yves Moëlo, the Associate Editor David Hibbs and Principal Editor Stuart Mills are greatly appreciated. The study was financially supported by the Ministry of Culture of the Czech Republic (long-term project DKRVO 2024-2028/1.II.a; National Museum, 00023272) for JS, PŠ and ZD.
Supplementary material
The supplementary material for this article can be found at https://doi.org/10.1180/mgm.2024.48.
Competing interests
The authors declare none.














 Open access
Open access