


dl-2-Hydroxy-(4-methylthio)butanoic acid (HMTBA) is a source of dietary l-methionine (Met) that is widely used in poultry nutrition and is transported across the apical membrane of enterocytes by a non-stereospecific, Na+-independent(Reference Brachet and Puigserver1) and H+-dependent carrier-mediated mechanism(Reference Maenz and Engele-Schaan2–Reference Martín-Venegas, Rodríguez-Lagunas and Geraert4), in addition to passive diffusion. The results obtained in a previous study in Caco-2 cells indicate the interaction of HMTBA with the H+-dependent monocarboxylate transporter 1 system(Reference Martín-Venegas, Rodríguez-Lagunas and Geraert4). Moreover, we have observed that monocarboxylate transporter 1 shows functional cooperation with the apical Na+–H+ exchanger-3, both in Caco-2 cells(Reference Martín-Venegas, Rodríguez-Lagunas and Geraert4) and in the chicken intestine(Reference Martín-Venegas, Geraert and Ferrer5). Monocarboxylate transporter 1 is a member of the proton-linked monocarboxylate transporter family, which mediates the transport of monocarboxylates such as lactate, pyruvate, ketone bodies and also SCFA(Reference Ritzhaupt, Wood and Ellis6, Reference Halestrap and Meredith7).
Upon absorption, the biological utilisation of HMTBA relies on its conversion to l-Met (Fig. 1), a process that starts in the intestine(Reference Martín-Venegas, Geraert and Ferrer8). The conversion of this synthetic source to the biologically active amino acid involves two enzymatic steps. In the first step, HMTBA is oxidised to 2-keto-(4-methylthio)butanoic acid (KMB) through a stereospecific enzymatic process. l-HMTBA is a substrate of the enzyme l-2-hydroxy acid oxidase (l-HAOX), which is present mainly in hepatic(Reference Dibner and Knight9, Reference Dibner and Ivey10) and renal(Reference Gordon and Sizer11) peroxisomes of chickens and rats(Reference Langer12). At least two isoforms of this enzyme have been described: isozymes A and B, the short-chain and long-chain hydroxyacid oxidases(Reference Duley and Holmes13). d-HMTBA is oxidised by the enzyme d-2-hydroxy acid dehydrogenase (d-HADH), which is found in the mitochondrial fraction of various tissues(Reference Knight and Dibner14). Subsequent to the formation of the common intermediate KMB, the second step is its conversion to l-Met by transamination, which is ubiquitous(Reference Gordon and Sizer11, Reference Knight and Dibner14–Reference Rangel-Lugo and Austic16).
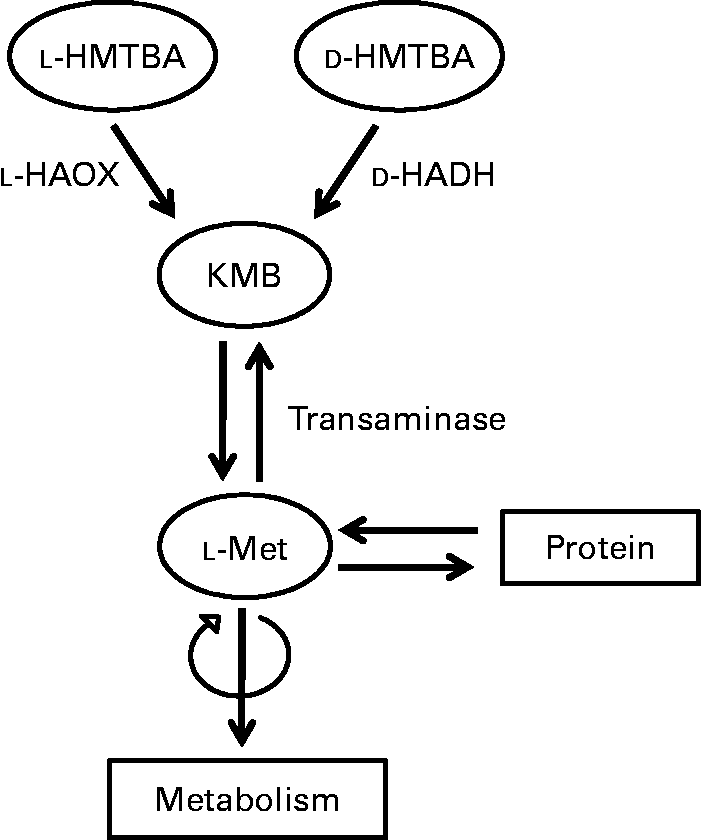
Fig. 1 Schematic illustration of the conversion of dl-2-hydroxy-(4-methylthio)butanoic acid (HMTBA) to l-methionine (l-Met). l-HAOX, l-2-hydroxy acid oxidase; d-HADH, d-2-hydroxy acid dehydrogenase; KMB, 2-keto-(4-methylthio)butanoic acid.
Given the importance of this Met source in animal production, knowledge of the contribution of the intestinal epithelium to the utilisation of HMTBA as a Met source would be of crucial interest. However, studies on the effect of dietary HMTBA on the conversion process are lacking. In the present study, we examined in differentiated intestinal Caco-2 cell cultures, HMTBA conversion to l-Met and further incorporation into cellular proteins as well as the regulation of HMTBA metabolism by HMTBA supplementation.
Materials and methods
Materials
Dulbecco's modified Eagle's medium, trypsin, penicillin and streptomycin were supplied by Gibco (Paisley, Scotland, UK). Non-essential amino acids, bovine serum albumin, fetal bovine serum, sterile PBS, d-glucose, HEPES, 2-(N-morpholino)ethanesulfonic acid, 2,6-dichlorophenolindophenol, p-toluenesulfonyl chloride, glycolic acid, 2-hydroxyisocaproic acid and d-lactate (Li+ salt) were supplied by Sigma (St Louis, MO, USA). Tissue culture supplies, including Transwells and clusters, were obtained from Costar (Cambridge, MA, USA). Unlabelled HMTBA was kindly provided by Adisseo France SAS (Antony, France). [1-14C]HMTBA (specific activity 2·035 GBq/mmol), [1-14C]l-Met (specific activity 2·035 GBq/mmol) and d-[2-3H]mannitol (specific activity 1·1 GBq/mmol) were purchased from ARC (St Louis, MO, USA). Filtron-X was supplied by National Diagnostics (Hessle, York, UK).
Cell culture
Caco-2 cells were kindly provided by Dr David Thwaites at the School of Cell and Molecular Biosciences, University of Newcastle upon Tyne (Newcastle upon Tyne, Tyne and Wear, UK) and cultured following Thwaites et al. (Reference Thwaites, Hirst and Simmons17), as described previously(Reference Martín-Venegas, Rodríguez-Lagunas and Geraert4). The cells (passages 117–122) were routinely grown in plastic flasks at a density of 5 × 104 cells/cm2 and cultured in Dulbecco's modified Eagle's medium containing d-glucose (4·5 g/l) and 2 mm-l-glutamine, and supplemented with 1 % (v/v) non-essential amino acids, 10 % (v/v) heat-inactivated fetal bovine serum, penicillin (100 IU/ml) and streptomycin (100 μg/ml) (control medium, 0·2 mm-l-Met) at 37°C in a modified atmosphere of 5 % CO2 in air. Cells maintained with 2 mm-HMTBA were cultured in Dulbecco's modified Eagle's medium with the same composition as the control medium but lacking l-Met (Gibco) and supplemented with 2 mm-HMTBA. This supplemented medium was filter-sterilised (0·2 μm membrane filter) before use.
All experiments were performed in cultures 19–21 d after seeding the cells in clusters (5 × 104 cells/cm2) for cell viability experiments; in filters (Transwells, pore size 0·4 μm, 12 mm in diameter, 4 × 105 cells/cm2) for transepithelial electrical resistance (TER), unidirectional d-mannitol fluxes and incorporation into cellular protein experiments; and in plastic flasks (5 × 104 cells/cm2) for d-HADH, l-HAOX and transamination evaluation.
Cell viability was estimated by ethidium bromide/acridine orange staining(Reference Parks, Bryan and Oi18) of cell suspensions after trypsin treatment, and is expressed as the number of viable cells × 103/cm2 of the monolayer.
Protein assay
Protein concentration was determined in cell homogenates by the Bradford method using the Bio-Rad protein assay kit, with bovine serum albumin as the standard. For monolayers grown on filters, the cultures were previously washed in PBS and incubated overnight in 750 μl of 0·5 m-NaOH.
Epithelial barrier function
Epithelial barrier function was estimated from TER and d-mannitol apical-to-basolateral fluxes, as described previously(Reference Martín-Venegas, Roig-Pérez and Ferrer19). Monolayers grown on filters were gently washed by sequential transfer through four beakers containing 500 ml of modified Krebs buffer (room temperature) with the following compositions: 137 mm-NaCl, 5·4 mm-KCl, 2·8 mm-CaCl2, 1·0 mm-MgSO4, 0·3 mm-NaH2PO4, 0·3 mm-KH2PO4, 10 mm-d-glucose and 10 mm-HEPES–Tris (pH 7·4). The monolayers were then placed in twelve-well clusters containing 1·5 and 0·70 ml of modified Krebs buffer in the basal and apical compartments, respectively. TER was determined using a Millicell-ERS voltohmmeter (Millipore, Bedford, MA, USA) and is expressed as Ω × cm2 of the monolayer surface area. The resistance of the supporting membrane was subtracted from all readings before calculations.
After TER determination, the apical medium was replaced by the same volume of modified Krebs buffer containing 137 mm-NaCl, 5·4 mm-KCl, 2·8 mm-CaCl2, 1·0 mm-MgSO4, 0·3 mm-NaH2PO4, 0·3 mm-KH2PO4, 10 mm-d-glucose, 0·1 mm-d-mannitol, 10 mm-2-(N-morpholino)ethanesulfonic acid–Tris (pH 5·5) and d-[2-3H]mannitol 7400 Bq/ml. Monolayers were incubated for 5 min at 37°C. At the end of the incubation, a sample of the basal medium was dissolved in 4 ml scintillation cocktail (Filtron-X) in preparation for counting in a Packard 1500 Tri-Carb counter (Downers Grove, IL, USA). The results are expressed as fmol/cm2.
d-2-Hydroxy acid dehydrogenase assay
Enzymatic activity was estimated following Schreiner & Jones(Reference Schreiner and Jones20) in samples prepared as described by Cammack(Reference Cammack21). After trypsinisation, cells were centrifuged (12 000 g, 5 min), and the pellet (50 mg protein) was resuspended with 3 ml of 1 mm-p-toluenesulfonyl chloride in 50 mm-PBS (pH 7·4) and homogenised for 2 min at 4°C in a glass with a Teflon pistil. The homogenate was transferred to a polypropylene tube and subjected to ultrasonic oscillation for 1 min with a Soniprobe (Dawe Instruments Limited, Brentford, Middlesex, UK) with a 12·7 mm tip. The output was adjusted to give a meter reading of 3 A, maintaining the samples below 0°C. Samples were then centrifuged (100 000 g, 75 min at 4°C). After centrifugation, the tubes were handled gently to avoid disturbance of the fatty layers at the top and bottom. Samples of 2·5 ml of the clear supernatants were removed with a syringe and applied to the top of a 10 × 1·1 cm column of Sephadex G-25 (Amersham International, South Glamorgan, Wales, UK) equilibrated with 25 mm-PBS, pH 6·3, at room temperature and recollected in different fractions of about 500 μl. The enzymatic assay buffer contained 200 μl of the purified protein (selected from the fractions with the highest protein concentration) and 1 ml of 300 μm-Tris–HCl buffer (pH 8·6), 56 nmol 2,6-dichlorophenolindophenol and 40 μmol d-lactate as a suitable substrate for d-HADH(Reference Cammack21, Reference Tubbs and Greville22). Because 2,6-dichlorophenolindophenol can be non-enzymatically reduced, a blank rate with no substrate was obtained for every reaction and subtracted from the identical assay containing the substrate. A unit of activity was defined as 1 nmol substrate oxidised/min, assuming E 600 = 21 000 /M per cm for 2,6-dichlorophenolindophenol. The results are expressed as mU/μg protein.
l-2-Hydroxy acid oxidase assay
Enzymatic activity was estimated following Schreiner & Jones(Reference Schreiner and Jones20). The cell monolayer was detached from the flask using a cell scraper and then centrifuged (3220 g, 5 min). The pellet was resuspended in 5 ml of 0·5 mm-Tris–HCl buffer (pH 7·4), 0·1 mm-EDTA and 1 mm-p-toluenesulfonyl chloride and homogenised for 2 min at 4°C in a glass with a Teflon pistil. Samples were then centrifuged (3220 g, 10 min), and the supernatants were removed and used for the assay. The enzymatic assay buffer (1·5 ml) contained 50 mm-Tris–HCl buffer (pH 8·0), 0·5 mm-sodium azide, 0·315 mm-o-dianisidine chlorhydrate, peroxidase (6 IU/ml), substrate (40 mmol/l) and 100 μl cell supernatant. The substrates tested were HMTBA and glycolic acid or l-2-hydroxyisocaproic acid as substrates of isozymes A and B, respectively(Reference Duley and Holmes13). Absorbance was measured at 436 nm every 30 s for 10 min at 30°C. A unit of activity was defined as 1 nmol substrate oxidised/min, assuming E 436 = 11·6 × 10− 3 /M per cm for o-dianisidine. The results are expressed as mU/μg protein.
Transamination study
Cells were gently washed in PBS, detached from the flask using a cell scraper and stored at − 80°C until the day of the enzymatic assay. Transamination experiments were performed following Rangel-Lugo & Austic(Reference Rangel-Lugo and Austic16). At the time of the experiment, samples were thawed, and Triton X-100 was added to achieve a detergent concentration of 0·1 % (w/v). The assay medium contained 1 mm-pyridoxal phosphate, 100 mm-d-mannitol, 44 mm-sucrose, 1 mm-dithiothreitol, 125 mm-HEPES, 16 mm-potassium phosphate, 2 mm-KMB and 2 mm-l-amino acid. The reaction was started by adding 100 μl of the cell sample (approximately 2 mg protein) to 400 μl of the assay media. Blanks for each l-amino acid tested lacked KMB. The reaction mixture was incubated for 40 min at 37°C in a shaking water-bath (70 oscillations/min), the assay was terminated by adding 100 μl of 10 % (w/v) sulfosalicylic acid, and the samples were then centrifuged (1000 g, 5 min). For quantification, 50 μl of 2 mm-norleucine were added to the supernatant (450 μl), and the sample was filtered by centrifugation (13 000 g, 15 min) using an Ultrafree® filter. Intracellular Met was quantified by ion exchange chromatography as described previously(Reference Martín-Venegas, Geraert and Ferrer8). The results are expressed as nmol/μg protein.
Incorporation of radioactivity into cellular proteins
After TER determination, monolayers grown on filters were incubated at 37°C in modified Krebs buffer (pH 5·5) containing 7400 Bq/ml of [1-14C]HMTBA or [1-14C]l-Met and 100 μmol/l of an unlabelled substrate in the apical compartment. After 30 min incubation, the filters were washed in modified Krebs buffer (pH 7·4, 4°C), removed from the insert and incubated for 1 h at 4°C in 500 μl of 10 % (v/v) TCA. The TCA-insoluble fraction, representing amino acid incorporation into newly synthesised proteins, was separated from the soluble fraction by centrifugation (15 000 g, 15 min, 4°C). A sample of the supernatant was taken for quantification of radioactivity, and the insoluble fraction was dissolved in scintillation cocktail for counting. A sample of the basolateral medium was also taken for quantification of radioactivity. Results are expressed as the percentage of radioactivity incorporated into the TCA-insoluble fraction with respect to the total radioactivity transported by the cells.
Statistical analysis
Results are given as means with their standard errors. Significant differences were detected by one-way ANOVA followed by Student's t test using SPSS® software (SPSS, Inc., Chicago, IL, USA). P < 0·05 was considered to denote significance.
Results
Since all the variables were tested in cultures maintained both in control conditions and in a medium containing 2 mm-HMTBA as a Met source, cell viability and epithelial barrier function were determined in cultures maintained in these conditions. The results (Table 1) indicate that neither cell number nor TER or d-mannitol fluxes were affected by the composition of the culture medium.
Table 1 Number of viable cells, transepithelial electrical resistance (TER) and d-mannitol fluxes in cells maintained under control conditions or in dl-2-hydroxy-(4-methylthio)butanoic acid (HMTBA)-supplemented medium (2 mmol/l)*
(Mean values with their standard errors, n 10)

* Mean values were not significantly different between the control and HMTBA-supplemented medium (P>0·05).
The first step in HMTBA conversion to l-Met is the stereospecific oxidation of HMTBA to KMB by d-HADH and l-HAOX. The results indicate that Caco-2 cells show d-HADH activity, which in turn is enhanced in HMTBA-maintained cells (control, 3·4 (sem 0·6) and HMTBA, 5·9 (sem 0·7), n 4, P < 0·05). As for l-HAOX, we tested glycolic acid and l-2-hydroxyisocaproic acid as the preferred substrates for isozymes A and B, respectively(Reference Duley and Holmes13), in addition to HMTBA. In control cells, the results show lower activity when 2-hydroxyisocaproic acid was used as a substrate, and no differences were observed between glycolic acid and HMTBA as substrates (Fig. 2). In contrast, in HMTBA-maintained cells, the results show no significant differences between the substrates and higher values than those detected under control conditions for all the substrates tested.
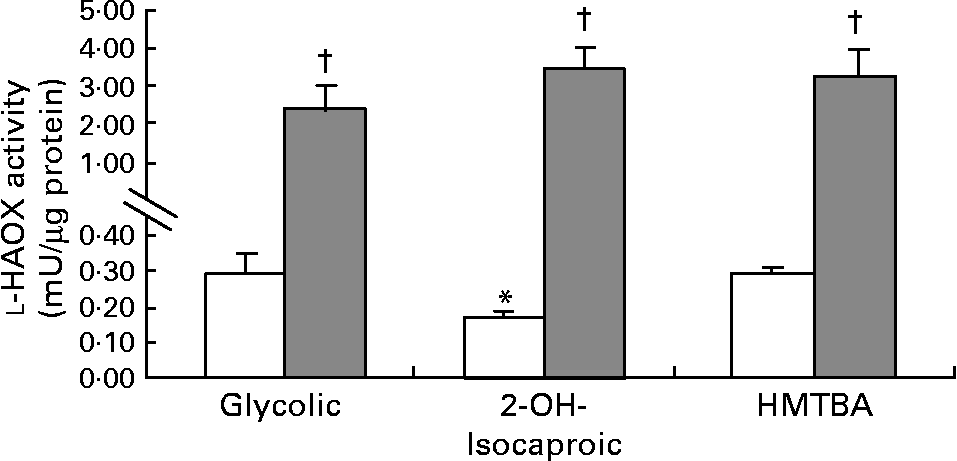
Fig. 2 l-2-Hydroxy acid oxidase (l-HAOX) activity in Caco-2 cells maintained in control (□) or in a medium supplemented with 2 mm-dl-2-hydroxy-(4-methylthio)butanoic acid (HMTBA; ![]() ). The enzymatic activity was estimated using glycolic acid (Glycolic), 2-hydroxyisocaproic acid (2-OH-Isocaproic) and HMTBA as substrates. Values are means, with standard errors represented by vertical bars (n 6). Mean values were significantly different between the substrates (*P < 0·05) and between the control and HMTBA-maintained cells for the same substrate (†P < 0·05).
). The enzymatic activity was estimated using glycolic acid (Glycolic), 2-hydroxyisocaproic acid (2-OH-Isocaproic) and HMTBA as substrates. Values are means, with standard errors represented by vertical bars (n 6). Mean values were significantly different between the substrates (*P < 0·05) and between the control and HMTBA-maintained cells for the same substrate (†P < 0·05).
The results obtained in the second step of HMTBA conversion, KMB transamination, indicate that all the amino acids tested can participate in the formation of l-Met (Fig. 3). The branched-chain amino acid l-leucine (Leu) is the most effective amino group donor, followed by l-glutamine, l-valine, l-alanine, l-phenylalanine and l-glutamic acid; lower l-Met formation was detected with l-proline, l-asparagine, l-serine, l-arginine and l-lysine followed by l-tyrosine and, finally, by l-aspartic acid. The conversion of KMB to l-Met was also studied in cells maintained under control conditions and in HMTBA-supplemented medium using l-Leu as the amino group donor in the enzymatic assay, with no significant differences in the levels of the amino acid obtained.
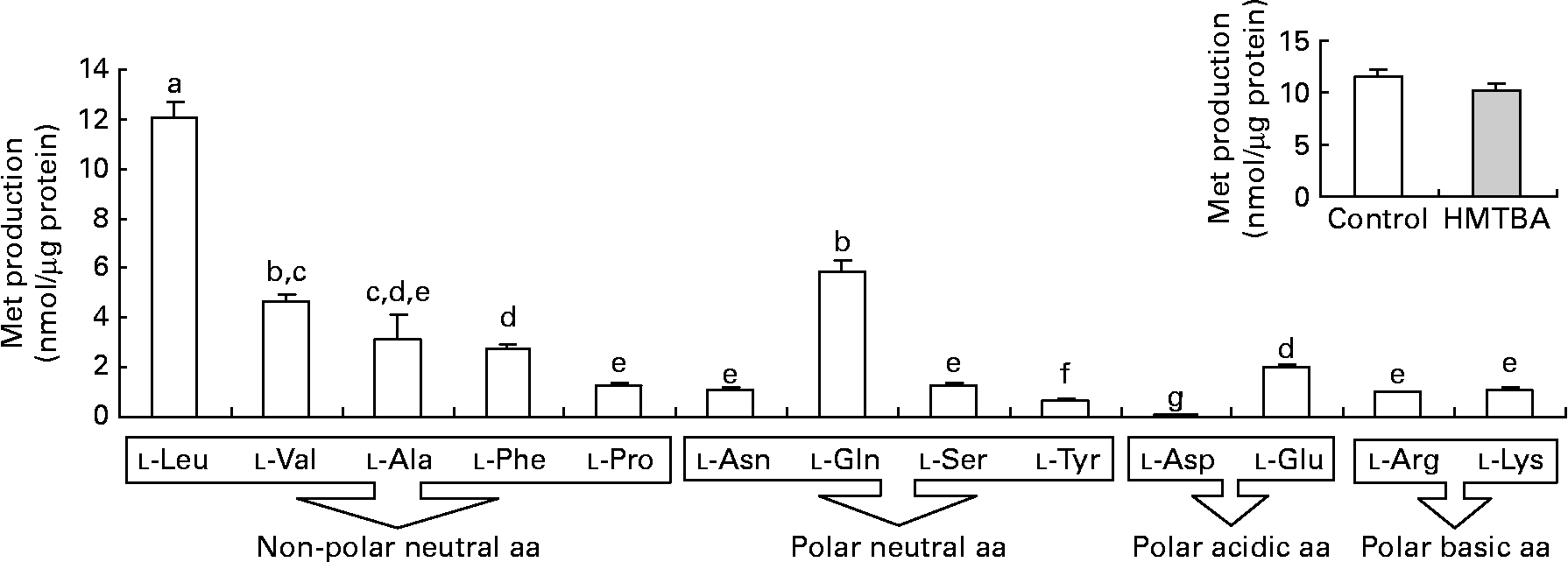
Fig. 3 Transamination of 2-keto-(4-methylthio)butanoic acid (KMB) to l-methionine (Met) in the presence of different amino acids (aa) as the amino group donors in Caco-2 cells maintained under control conditions. Inset: KMB conversion to l-Met in cells maintained under control or in a medium supplemented with 2 mm-dl-2-hydroxy-(4-methylthio) (□) butanoic acid (HMTBA; ![]() ) in the presence of l-Leu as the amino group donor. Values are means, with standard errors represented by vertical bars (n 4). a,b,c,d,e,f,g Mean values with unlike letters were significantly different (P < 0·05). Leu, leucine; Val, valine; Ala, alanine; Phe, phenylalanine; Pro, proline; Asn, asparagine; Gln, glutamine; Ser, serine; Tyr, tyrosine; Asp, aspartic acid; Glu, glutamic acid; Arg, arginine; Lys, lysine.
) in the presence of l-Leu as the amino group donor. Values are means, with standard errors represented by vertical bars (n 4). a,b,c,d,e,f,g Mean values with unlike letters were significantly different (P < 0·05). Leu, leucine; Val, valine; Ala, alanine; Phe, phenylalanine; Pro, proline; Asn, asparagine; Gln, glutamine; Ser, serine; Tyr, tyrosine; Asp, aspartic acid; Glu, glutamic acid; Arg, arginine; Lys, lysine.
The incorporation of HMTBA radioactivity into cellular proteins was quantified as an indicator of HMTBA conversion to l-Met. In control cells, there were no significant differences between the incorporation of HMTBA and l-Met radioactivity in the TCA-insoluble fraction (Table 2). The results also indicate that incorporation of HMTBA radioactivity was not affected by HMTBA supplementation.
Table 2 Incorporation of l-methionine (Met) and dl-2-hydroxy-(4-methylthio)butanoic acid (HMTBA) radioactivity into the cell protein of cultures maintained under control conditions or in HMTBA-supplemented medium (2 mmol/l))*
(Mean values with their standard errors, n 4)
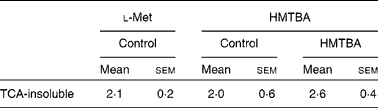
* Incorporation of radioactivity into the TCA-insoluble fraction was estimated after 30 min incubation of the cells with the labelled substrate in the apical compartment. Results are expressed as a percentage of radioactivity incorporated into the TCA-insoluble fraction with respect to the total radioactivity accumulated in the cells. Mean values were not significantly different between either the labelled substrates or the control and HMTBA-supplemented medium (P>0·05).
Discussion
To investigate the effects of supplementation of the culture medium with HMTBA, it was first necessary to test specific indicators of culture progression. Cultures maintained in filters are normally seeded at a high cell density to ensure that all the cells achieve confluence at the same time, so that the expression of metabolic enzymes is homogeneous(Reference Le Ferrec, Chesne and Artusson23). The present results indicate that cell number was not affected by HMTBA supplementation. Moreover, the integrity of the monolayer assessed from TER and d-mannitol fluxes confirms these data. Therefore, the differences observed cannot be attributed to changes in either the viability or the differentiation state of the epithelium.
In terms of oxidation of this Met precursor, the present results indicate that Caco-2 cells can convert d-HMTBA to KMB. Moreover, d-HADH activity increases by approximately twofold when HMTBA availability is enhanced. These results suggest up-regulation of this enzyme by the dietary content. As for l-HAOX, the present results also suggest the activity of this enzyme in Caco-2 cells. Moreover, the data indicate that both l-HAOX-A and -B isozymes are present. To our knowledge, this is the first time that the presence of both isozymes in the intestinal epithelium has been reported. In control cells, the highest activity was detected on glycolic acid, thus suggesting that isozyme A may be more active. In HMTBA-maintained cells, the activity of both isoforms clearly increased, thus indicating, as for d-HADH, a dietary up-regulation effect. Robinson et al. (Reference Robinson, Keay and Molinari24) suggested that l-HMTBA might be a more suitable substrate for isozyme B than for isozyme A, and reported the presence of both isozymes in the hog renal cortex. Nevertheless, Schreiner & Jones(Reference Schreiner and Jones20) partially purified the enzyme in this tissue and observed a single form that was able to oxidise glycolic acid and, to a lesser extent, l-HMTBA and l-mandelic acid. Dupuis et al. (Reference Dupuis, Brachet and Puigserver25) reported the presence of both isozymes in the chicken liver and 3 years later, in the chicken kidney(Reference Dupuis, De Caro and Puigserver26), and concluded that both were probably involved in l-HMTBA oxidation, although isozyme B has a higher affinity for this Met source. In control cells, we observed the highest activity on glycolic acid and HMTBA, thus suggesting that HMTBA is preferentially oxidised by isozyme A unless isoform B has a higher affinity for HMTBA than for l-2-hydroxyisocaproic acid. Moreover, in HMTBA-supplemented conditions, there were no differences between the substrates, indicating a similar increase in glycolic acid and HMTBA (nine- and thirteenfold increase, respectively) and a more pronounced increase in l-2-hydroxyisocaproic acid (thirty-fivefold), thus suggesting that isozyme B is more sensitive to HMTBA availability. Therefore, the present results do not allow the identification of the isozyme responsible for l-HMTBA oxidation but indicate that Caco-2 cells express all the enzymes involved in the conversion of d- and l-HMTBA to KMB. Similarly, Schreiner & Jones(Reference Schreiner and Jones20) in porcine kidney fibroblasts and McCollum et al. (Reference McCollum, Vázquez-Añón and Dibner27) in isolated sheep ruminal and omasal epithelia also detected the capacity to convert d- and l-HMTBA to KMB. Moreover, the present study reveals that this conversion can be enhanced by nutritional HMTBA supplementation.
In the second step, KMB formed from both d- and l-HMTBA is transaminated to l-Met. In chickens, it is described that a wide variety of transaminases can catalyse this reaction(Reference Knight and Dibner14–Reference Rangel-Lugo and Austic16). Although the liver is the main organ in which HMTBA is converted to l-Met, it has been reported that other organs may also participate in this process(Reference Dibner and Knight9, Reference Dibner and Ivey10–Reference Langer12, Reference Dupuis, Brachet and Puigserver25, Reference McCollum, Vázquez-Añón and Dibner27). KMB is an intermediary metabolite in other metabolic reactions, but, in the chicken intestine, the formation of l-Met is the preferred pathway(Reference Brachet and Puigserver28). Moreover, a recent investigation in sheep has demonstrated that the gastrointestinal tract synthesises l-Met via transamination of dietary HMTBA(Reference Lobley, Wester and Calder29, Reference Lobley, Wester and Holtrop30). The present results indicate that KMB transamination to l-Met in Caco-2 cells is also possible, thus confirming the participation of the intestine in this enzymatic reaction. l-Leu was found to be the preferred amino group donor, although the results suggest that transamination is not linked to a specific amino group donor since all the amino acids tested promoted the formation of l-Met. Gordon & Sizer(Reference Gordon and Sizer11) suggested that branched-chain amino acids are the most preferred amino group donors in KMB transamination in the chicken liver. In contrast, in the rat intestine, it has been suggested that l-glutamine is the principal amino donor in transamination, although neither l-Leu nor other branched-chain amino acids were tested(Reference Langer12). In this sense, Rangel-Lugo & Austic(Reference Rangel-Lugo and Austic16) observed that of all the amino acids tested (l-isoleucine, l-Leu, l-valine, l-glutamic acid, l-aspartic acid, l-glutamine, l-asparagine and l-phenylalanine), l-Leu was the most effective amino group donor. In HMTBA-maintained cells, the formation of l-Met from KMB is not modified, thus confirming that transamination is not the limiting step in HMTBA conversion.
Upon l-Met formation from HMTBA, this nutritionally indispensable amino acid is incorporated into many metabolic pathways: (1) protein synthesis; (2) transmethylation to form S-adenosylmethionine, a primary methyl donor that participates in the methylation process to form metabolites such as creatine and phosphatidylcholine, and also in polyamine synthesis; (3) trans-sulfuration to form l-cysteine, which in turn is also a precursor for protein synthesis and can be incorporated into glutathione or catabolised to taurine. In the present study, the incorporation of HMTBA radioactivity into cellular proteins was evaluated as an indicator of HMTBA conversion to l-Met. The present results indicate that the incorporation of radioactivity into cellular proteins is similar for labelled HMTBA and l-Met. Gordon & Sizer(Reference Gordon and Sizer11) found that the radioactivity of HMTBA and l-Met was equally incorporated into liver proteins and distributed in the blood plasma and excreta. Similarly, Dibner(Reference Dibner31) found no differences in the rate of incorporation of radioactivity from labelled HMTBA and Met into protein in isolated chicken hepatocytes, nor were there any differences in porcine hepatocytes incubated with HMTBA and dl-Met(Reference Knight, Atwell and Wuelling32). Saunderson(Reference Saunderson33) found that although incorporation of radioactivity from HMTBA, dl-Met or l-Met into proteins is similar in the liver and kidney, they behave differently from other tissues since skeletal muscle, heart, skin and brain all show much lower incorporation from HMTBA and dl-Met than from l-Met. Therefore, the present results suggest that the intestine would behave in a similar manner to the liver and kidney. Finally, the results also show that there was no difference in the percentage of radioactivity incorporated into proteins in the control and HMTBA-supplemented cells.
In summary, the results obtained for d-HADH, l-HAOX and transamination experiments in Caco-2 cells suggest that the complete conversion of d- and l-HMTBA to l-Met takes place in the intestinal epithelium. Moreover, HMTBA supplementation enhances intestinal oxidation to KMB, thus increasing the availability of l-Met immediately upon absorption. KMB transamination is not specifically linked to an amino group donor and does not constitute the limiting step in HMTBA conversion. Finally, the data also indicate that the conversion of HMTBA does not limit the incorporation of l-Met into protein. In conclusion, the results reveal the capacity of Caco-2 cells to convert HMTBA to l-Met and the up-regulation of conversion by dietary HMTBA, thus highlighting the contribution of the intestinal epithelium in the utilisation of HMTBA as a source of Met. Although the translation of in vitro Caco-2 cell data into an in vivo situation necessitates further investigation, this model is a useful tool for the appraisal of intestinal metabolism, and, therefore, the results obtained could constitute the basis for future dietary strategies designed to improve chicken performance.
Acknowledgements
The present study was supported by Fundació Bosch i Gimpera and Adisseo France SAS. The authors have no conflicts of interest. R. M.-V., Y. M., P.-A. G. and R. F. designed the research (project conception, development of overall research plan and study oversight), R. M.-V. and M. T. B. conducted the experiments and the statistical analysis. R. M.-V. and R. F. wrote the manuscript, and all the authors had responsibility for the final content. All authors read and approved the final manuscript. The authors thank Dr Josefina Prat for her guidance and advice in spectrophotometric techniques. The valuable help of the Serveis Cientificotècnics of the Universitat de Barcelona is also gratefully acknowledged.





