Humans and other mammals maintain their core body temperature (Tc) at 37°C to ensure optimal physiological function and survival. Prolonged exposure to heat can raise Tc and result in heat-related illnesses or even death(Reference Williams1). The global temperature record of the last 2000 years shows the largest increase (1.09°C above pre-industrial times) in the 2011–2020 decade, and leading climate scientists predict the world to warm more than 3°C by the end of the century(Reference Tollefson2). As of today, ∼30 % of the world’s population is exposed to deadly heat waves for at least 20 days per year(Reference Mora, Dousset and Caldwell3). During the latest June 2021 heat wave in the USA, the Pacific Northwest Coast alone reported 3504 heat-related illness emergency department visits(Reference Schramm, Vaidyanathan and Radhakrishnan4). Global warming is indeed a threat to human health and survival. The development of effective prevention and treatment strategies for heat-related illness and injuries is therefore crucial.
Mitochondria are double membrane-bound organelles that produce cellular energy and coordinate stress responses (e.g., to heat) for determining cell life-or-death(Reference Yu, Wang and Yoon5). The overall mitochondrial morphology is determined by the balance between mitochondrial fission and fusion, which is regulated by the dynamin family of large GTPases(Reference Yu, Wang and Yoon5–Reference Willems, Rossignol and Dieteren7). In mammals, the cytosolic protein Drp1 is recruited to the mitochondria to promote mitochondrial fission(Reference Yu, Wang and Yoon5,Reference Yu, Fox and Burwell8) . Mitofusins (Mfn1 and Mfn2) on the outer mitochondrial membrane and OPA1 on the inner mitochondrial membrane coordinate the fusion of their respective membranes(Reference Chen, Detmer and Ewald9,Reference Del Dotto, Fogazza and Carelli10) . Heat exposure activates the mitochondrial fission protein Drp1 and degrades the fusion protein OPA1, which results in mitochondrial fragmentation and dysfunction(Reference Yu, Ferdjallah and Elenberg11–Reference Sanjuan Szklarz and Scorrano13). Mitochondrial fission may thus serve as a therapeutic target for heat-related illness, as inhibition of mitochondrial fission restores mitochondrial function under heat stress and ameliorates heat-induced oxidative damage to skeletal muscle(Reference Yu, Ferdjallah and Elenberg11,Reference Yu, Deuster and Chen12) .
l-Citrulline is a naturally occurring α-amino acid which is found in foods such as watermelon. l-Citrulline is absorbed by the enterocytes in the small intestine and converted in the kidneys to l-arginine, a biological precursor of nitric oxide (NO)(Reference Allerton, Proctor and Stephens14). Supplementation with l-citrulline has been shown to increase NO levels in plasma and tissues, including skeletal muscle(Reference Allerton, Proctor and Stephens14,Reference Ham, Gleeson and Chee15) . NO is a gaseous signalling molecule that plays a critical role in many physiological functions, including but not limited to vasodilation, antioxidant, anti-inflammatory and heat shock responses(Reference Allerton, Proctor and Stephens14,Reference Ghimire, Altmann and Straub16,Reference Uyanga, Wang and Tong17) . NO may directly or indirectly increase mitochondrial mass and stimulate mitochondrial biogenesis in skeletal muscle(Reference Tengan and Moraes18–Reference Tengan, Rodrigues and Godinho20). In neurons, NO induces S-nitrosylation (SNO) of the mitochondrial fission protein Drp1, which contributes to the pathogenesis of neurodegeneration(Reference Cho, Nakamura and Fang21). Reduced NO bioavailability in skeletal muscle has been associated with muscle wasting and impaired function(Reference Allerton, Proctor and Stephens14,Reference Ham, Gleeson and Chee15,Reference Tengan, Rodrigues and Godinho20) . Chowdhurya et al. have shown that chickens exposed to heat had significantly lower plasma l-citrulline concentrations and brain oxidative damage(Reference Chowdhury, Tomonaga and Ikegami22). However, the effects of l-citrulline and NO on heat-induced damage of mitochondria and skeletal muscle injury remain to be examined.
Skeletal muscle is highly susceptible to heat injury(Reference Chen and Yu23). Our recent studies suggest that mitochondrial fission mediates heat-induced reactive oxygen species (ROS) production and skeletal muscle injury(Reference Yu, Ferdjallah and Elenberg11,Reference Yu, Deuster and Chen12) . In this study, we tested whether l-citrulline would be effective in preventing heat-induced mitochondrial dysfunction and cell injury in mouse C2C12 myoblasts. Moreover, we wanted to elucidate the mechanisms responsible for the downstream effect of l-citrulline-induced NO production, with particular emphasis on mitochondrial fission machinery. Our results show that l-citrulline ameliorates heat-induced skeletal muscle oxidative injury through NO-mediated Drp1 inhibition and preservation of mitochondrial functional integrity.
Materials and methods
C2C12 myoblast culture, heat exposure and l-citrulline treatment
The mouse myoblast C2C12 cell line (ATCC® CRL-1772™) was cultured in Dulbecco’s Modified Eagle Medium supplemented with 10 % fetal bovine serum, 100 unit/ml penicillin and 100 μg/ml streptomycin. Cells were maintained at 37°C in a 5 % CO2 humidified incubator and at a confluency below 60 % to prevent differentiation. For heat exposure, the cells were placed in a CO2 incubator with a temperature preset to 43°C, as previously described(Reference Yu, Deuster and Chen12).
l-Citrulline was purchased from Sigma-Aldrich (#C7629, purity ≥ 98 % by TLC, Burlington, MA), dissolved in phosphate-buffered saline and added to the cell culture medium (2 mM) for the indicated time, or 1 h prior to heat exposure. Control cells were treated with phosphate-buffered saline only. The concentration of 2 mM citrulline can be achieved in plasma in humans 1 h after oral supplementation with l-citrulline(Reference Churchward-Venne, Cotie and MacDonald24), in mice with l-citrulline-injected intraperitoneally(Reference Wijnands, Meesters and van Barneveld25) or in rats via subcutaneous injection(Reference Vadivel, Aschner and Rey-Parra26). A previous study performed dose–response experiments in C2C12 cells and the results show that a similar concentration of l-citrulline (2.5 mM) protected muscle from wasting(Reference Ham, Gleeson and Chee15).
Assessment of mitochondrial morphology and membrane potential (ΔΨm)
Mitochondria were visualised using a cell-permeable probe MitoTracker Red CMXRos, and mitochondrial membrane potential (ΔΨm) was measured with the cationic fluorescent dye – tetramethylrhodamine ethyl ester. Both fluorescent dyes were purchased from Thermo Fisher Scientific and added to the cell culture medium (100 nM) 15 min prior to imaging, per the manufacturer’s instructions. Percentage of cells containing fragmented or elongated mitochondria was counted as previously described(Reference Yu, Robotham and Yoon27–Reference Yu, Dohl and Elenberg29). Using ImageJ software (NIH), mitochondrial morphology was further assessed as the form factor (perimeter2/4π·surface area) and aspect ratio (major axis/minor axis), which reflects the branching and length aspects of mitochondria, respectively. Since changes in mitochondrial shapes and volumes may alter fluorescence signal in the mitochondria, ΔΨm was calculated as mean tetramethylrhodamine ethyl ester fluorescence intensity per mitochondrial area.
Measurements of cellular levels of nitric oxide and reactive oxygen species
The cell-permeable nonfluorescent DAF-FM diacetate (Thermo Fisher Scientific) reacts with NO to form a fluorescent benzotriazole (DAF-FM). Cellular NO levels were determined by DAF-FM fluorescence intensity. Nω-nitro-l-arginine methyl ester hydrochloride (l-NAME, Sigma-Aldrich, 1 mM), a non-selective NO synthase inhibitor, was added to the cell culture medium to inhibit cellular NO signalling. ROS levels were detected by dihydroethidium (Thermo Fisher Scientific) following the manufacturer’s instructions.
Cell lysate, mitochondria fractionation, S-nitrosylation Drp1 detection and western blot
Cell lysate was obtained by incubating cells with ice-cold RIPA buffer containing protease inhibitors, and mitochondria fractionation was performed using the Thermo Scientific Mitochondria Isolation Kit for Cultured Cells per manufacturer’s instructions. Cell lysate or mitochondrial protein (∼10 μg) was loaded into polyacrylamide gels (Bio-Rad Laboratories) and separated by SDS-PAGE.
The following primary antibodies were used: mouse anti-Drp1 (Catalog #611112, BD Biosciences), mouse anti-OPA1 (#612 606, BD Biosciences), rabbit anti-MFN1 (#14 739) and anti-MFN2 (#9482, Cell Signaling Technology), rabbit anti-phospho-DRP1-Ser616 (#4494) and anti-phospho-DRP1-Ser637 (#6319, Cell Signaling Technology), rabbit anti-VDAC (#4661, Cell Signaling Technology) and rabbit anti-GAPDH (#ABS16, Sigma-Aldrich). Horseradish peroxidase-conjugated anti-rabbit and anti-mouse antibodies were used as secondary antibodies. Primary antibodies were used at 1:1000 dilution and secondary antibodies at 1:5000. SNO-Drp1 was detected using a Pierce S-Nitrosylation Western Blot Kit (Thermo Fisher Scientific) following the manufacturer’s instructions. Protein expression was visualised with Western ECL Blotting Substrates (Bio-Rad), and image acquisition was performed using a Bio-Rad ChemiDoc MP Imaging System. Densitometry was performed using ImageJ software (NIH).
Caspase 3/7 activities, cell apoptosis and cell viability assays
Caspase 3/7 activity was measured with the CellEventTM Caspase-3/7 Green Detection Reagent (Thermo Fisher Scientific) following the manufacturer’s instructions. Apoptosis was determined using an Annexin V Alexa Fluor® 488 apoptosis kit (Thermo Fisher Scientific). Cell viability was determined using the trypan blue exclusion test with a TC20 automated cell counter (Bio-Rad) per manufacturer’s instructions and confirmed by cell counting with a light microscope.
Fluorescence microscopy
Fluorescence images were viewed and acquired with a Nikon Eclipse Ti epifluorescence microscope equipped with a digital camera (Melville, NY). Excitation/emission wavelengths were 480/535 nm for green fluorescence (annexin V, caspase-3/7 Green and DAF-FM) and 555/613 nm for red fluorescence (MitoTracker red, tetramethylrhodamine ethyl ester and dihydroethidium). The images were analysed using ImageJ software (NIH).
Statistical analysis
All data are presented as mean ± SDs. Statistical analyses were performed using GraphPad Prism 9 Software. A one-way ANOVA with Tukey’s multiple comparisons test was used to compare mean group differences. For cell viability under heat stress, a repeated-measures ANOVA was used to determine the main effect of l-citrulline treatment. Values of P < 0·05 were considered statistically significant.
Results
l-Citrulline induces mitochondrial elongation under normal temperature
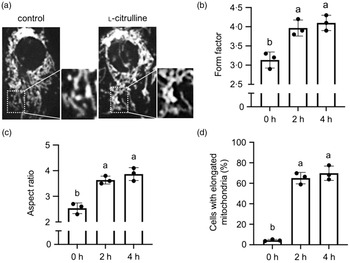
We first examined whether l-citrulline has an effect on mitochondrial morphology under normal temperature. As shown in Fig. 1(a), mitochondrial shape was variable among C2C12 myoblasts and consisted of small spheres, short rods and tubular filaments. In l-citrulline-treated cells, most mitochondria were elongated in a tubular form with interconnected reticular network (Fig. 1(a)). l-Citrulline treatment for 2 and 4 h increased both mitochondrial form factor (branching) and aspect ratio (length) in C2C12 myoblasts (Fig. 1(b) and (c)), and the percentages of cells containing elongated mitochondria increased by 60·7 % and 65·5 %, respectively (Fig. 1(d)). l-Citrulline treatment did not affect ΔΨm in cells at normal temperature (data not shown). Taken together, these data suggest that l-citrulline induces mitochondrial elongation under normal temperature.
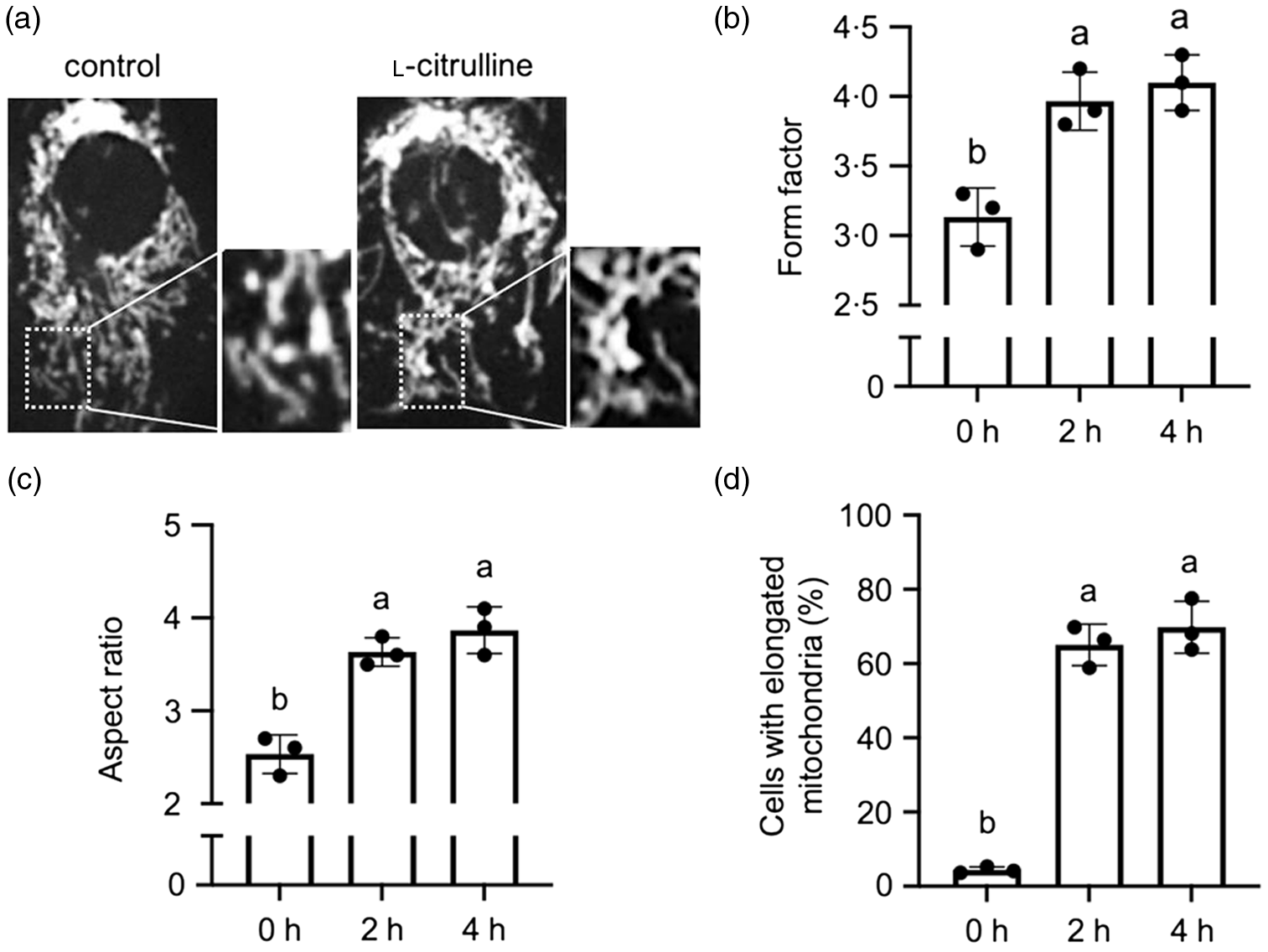
Fig. 1. l-Citrulline induces mitochondrial elongation under normal temperature. (A) Representative images of C2C12 myoblasts with mitochondria labelled by MitoTracker Red. Cells were incubated in phosphate-buffered saline (control) or 2 mM l-citrulline at 37°C for 2 h. Scale bar: 10 µm; inserts: 2 × magnification Quantification of mitochondrial (b) form factor (degree of branching) and (c) aspect ratio (length-to-width ratio) in cells treated with l-citrulline for 2 and 4 h. (d) Percentage of cells with elongated mitochondria. Values are means ± sd; n = 3 independent experiments; 20 cells (b) and (c) or ∼300 cells (d) were randomly selected for quantification in each group. Labelled means without a common letter differ, P < 0.05.
The effects of l-citrulline on mitochondrial fission and fusion machinery under normal temperature
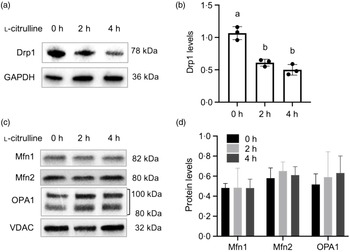
As depicted in Fig. 2(a) and (b), 2 and 4 h l-citrulline incubations decreased mitochondrial fission factor Drp1 protein expression in normal temperature cells. No alterations were found for the mitochondrial fusion proteins Mfn1, Mfn2 and OPA1 in cells treated with l-citrulline (Fig. 2(c) and (d)). These results suggest that l-citrulline induces mitochondrial elongation through fission inhibition
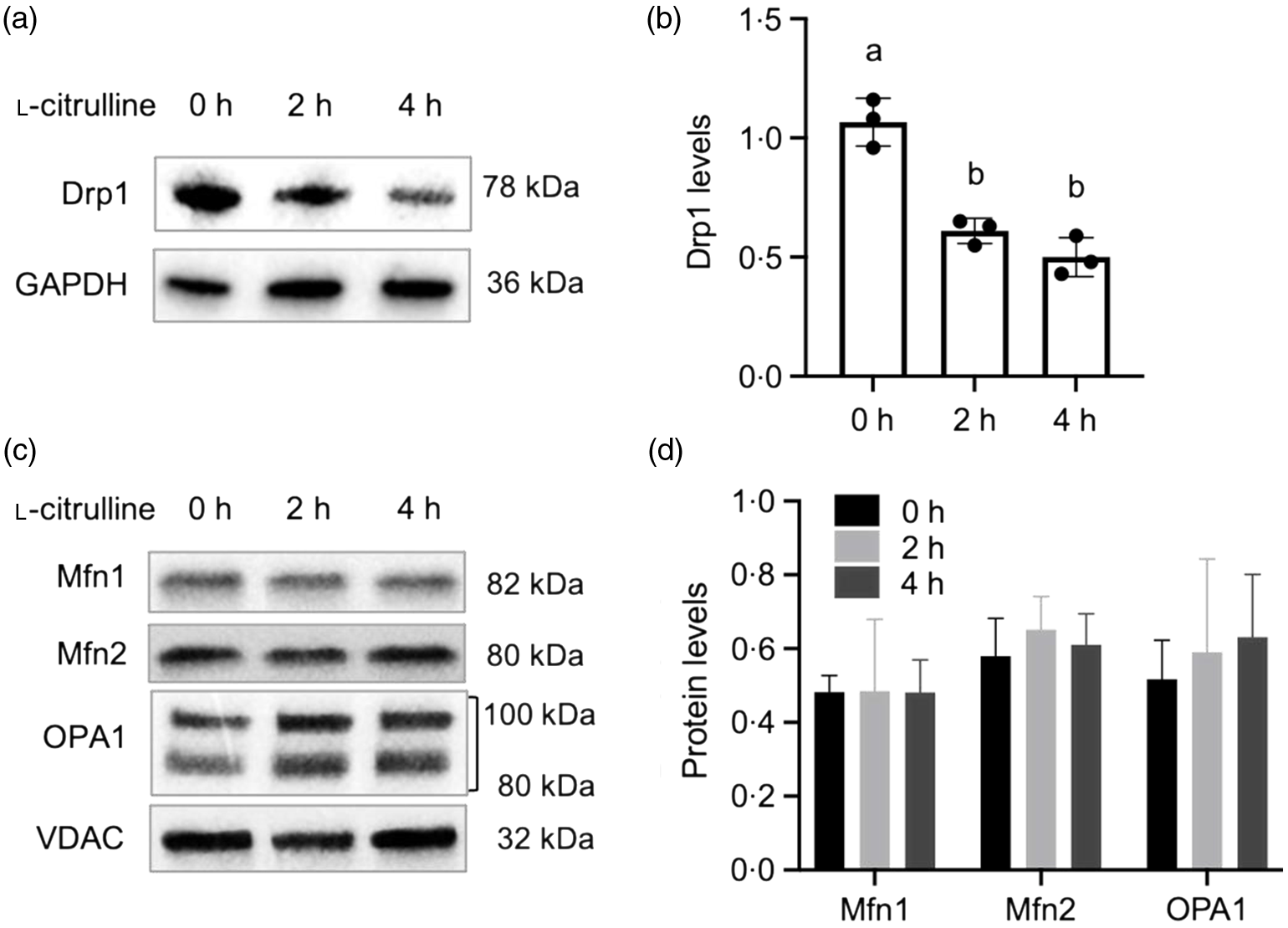
Fig. 2. The effects of l-citrulline on mitochondrial fission and fusion machinery under normal temperature. (a) Representative western blot images of mitochondrial fission protein Drp1. GAPDH served as a loading control. (b) Densitometry of Drp1 protein expression, values are relative to GAPDH. (c) Representative western blot images of mitochondrial fusion proteins Mfn1, Mfn2 and OPA1. VDAC served as a loading control. (d) Densitometry of mitochondrial fusion proteins expression (relative to VDAC). Values are means ± sd; n = 3 independent experiments. Labelled means without a common letter differ, P < 0.05.
l-Citrulline increases cellular nitric oxide levels, but not S-nitrosylation of Drp1 under normal temperature
l-Citrulline is the precursor of l-arginine and a substrate for NO synthase. Indeed, l-citrulline treatment increased cellular NO levels in C2C12 myoblasts (Fig. 3(a)). l-NAME, a non-selective NO synthase inhibitor, prevented the l-citrulline-induced increase in NO levels. Cellular ROS levels were not influenced by l-citrulline (Fig. 3(b)). Although l-citrulline promotes NO production, it did not increase SNO-Drp1 levels in C2C12 myoblasts (Fig. 3(c) and (d))
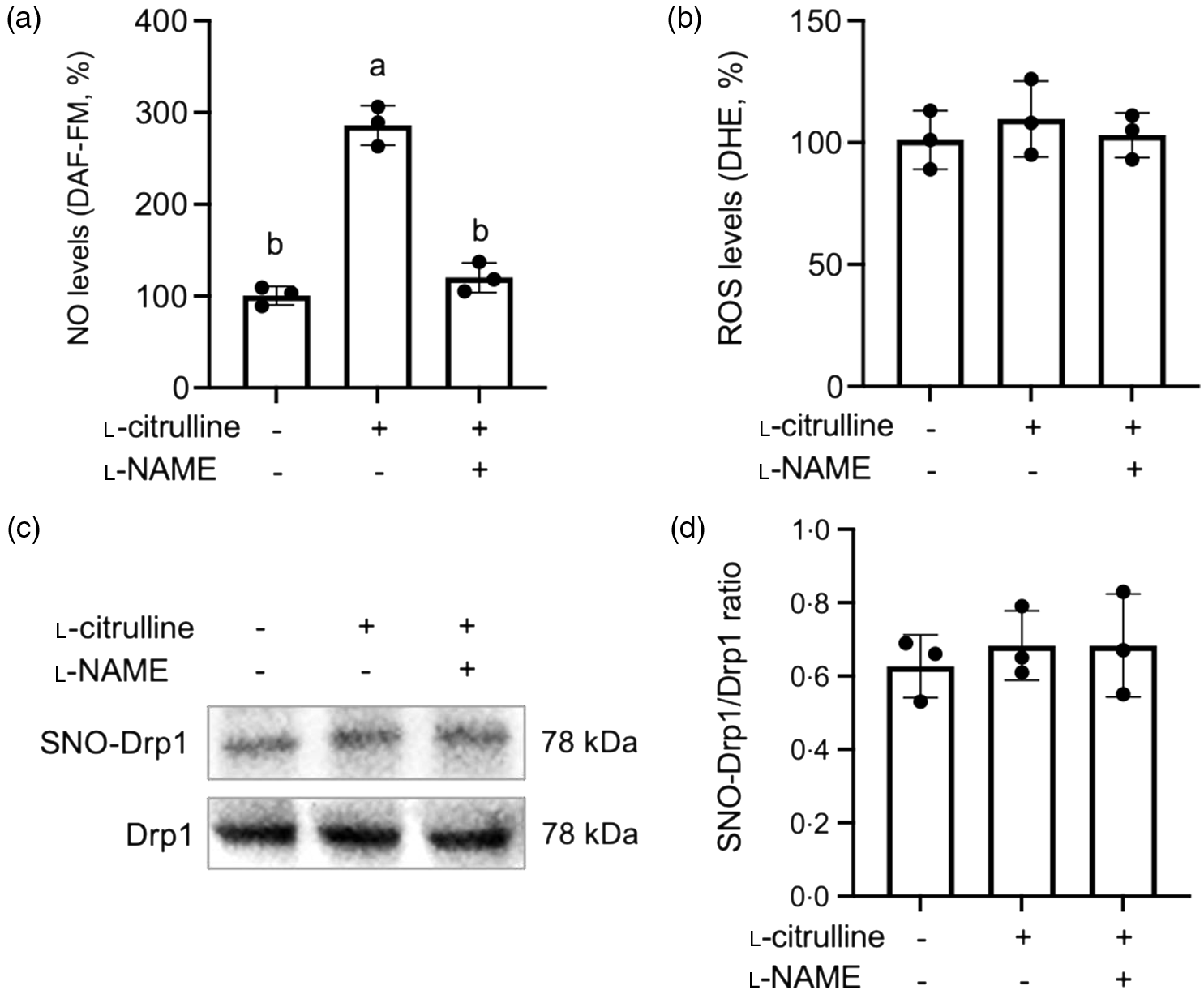
Fig. 3. l-Citrulline increases cellular nitric oxide (NO) levels but does not increase S-nitrosylation (SNO) of Drp1 under normal temperature. Cellular NO levels (a) and reactive oxygen species (ROS) levels (b) were determined by DAF-FM and dihydroethidium fluorescence intensity, respectively. Values are % to control cells. (C) Representative western blot images of SNO-Drp1 protein expression. Drp1 served as a loading control. (d) Densitometric analysis of SNO-Drp1 to Drp1 ratio. Cells were treated with phosphate-buffered saline (control) or 2 mM l-citrulline for 2 h; l-NAME (1 mM) is a non-selective NO synthase inhibitor. Values are means ± sd; n = 3 independent experiments. Thirty cells were randomly selected for quantification of NO and ROS levels in each group. Labelled means without a common letter differ, P < 0.05.
l-Citrulline inhibits mitochondrial fission by regulating Drp1 phosphorylation through elevated levels of nitric oxide under normal temperature
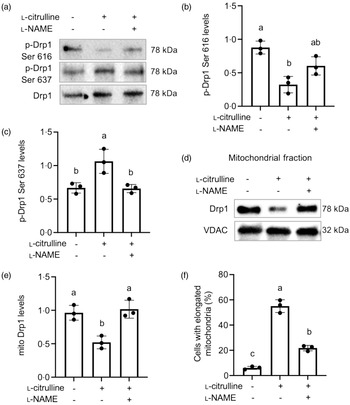
To outline the regulatory mechanism for l-citrulline-induced mitochondrial fission inhibition, we examined the phosphorylation status of Drp1 and its mitochondrial distribution. l-Citrulline caused a decrease in phosphorylation of Drp1 at Ser 616 and an increase in phosphorylation of Drp1 at Ser 637, both of which were blocked by l-NAME (Fig. 4(a)–(c)). Subcellular fractionation and localisation studies further revealed that l-citrulline reduced mitochondrial localisation of Drp1 (Fig. 4(d) and (e)). l-NAME abolished the l-citrulline-induced decrease in mitochondrial localisation of Drp1 and mitochondria elongation (Fig. 4(e) and (f)). Together, these results suggest that l-citrulline inhibits mitochondrial fission by regulating Drp1 phosphorylation through elevation in NO levels.
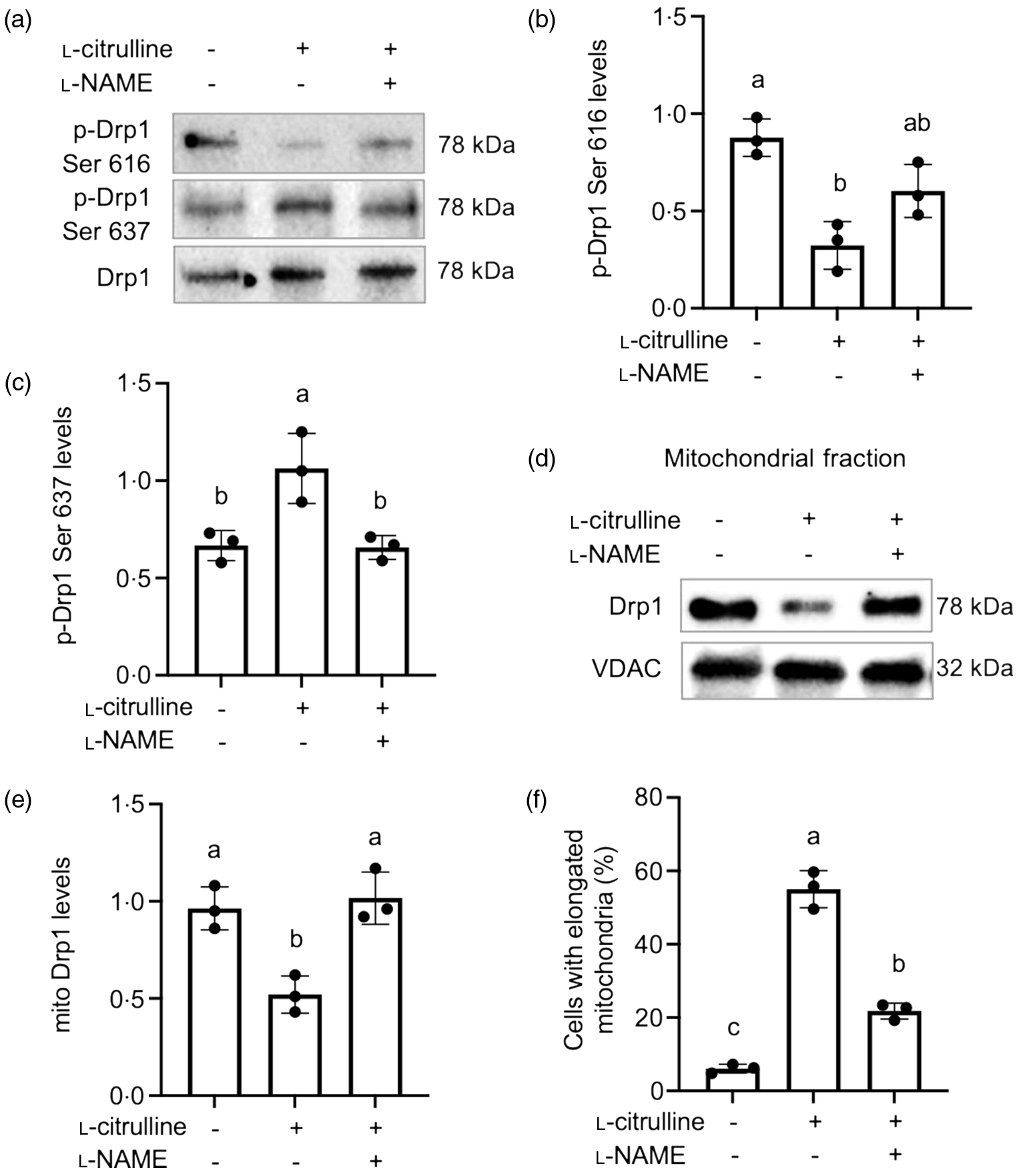
Fig. 4. l-Citrulline induced nitric oxide -dependent Drp1 inhibition under normal temperature. (a) Representative western blot images of phospho-Drp1 Ser 616 and phospho-Drp1 Ser 637; total Drp1 served as a loading control. Densitometry of phospho-Drp1 Ser 616 (b) and phospho-Drp1 Ser 637 (c) levels; values are ratio to total Drp1. (d) Representative western blot images of Drp1 in the mitochondrial fractions. VDAC served as a mitochondrial loading control. (e) Densitometric analysis of mitochondrial (mito) Drp1 levels (ratio to VDAC). Values are means ± sd; n = 3 independent experiments and ∼300 cells were counted in each group for mitochondrial morphology (f). Labelled means without a common letter differ, P < 0.05.
l-Citrulline prevents heat-induced translocation of Drp1 to mitochondria, mitochondrial fragmentation and decreased ΔΨm
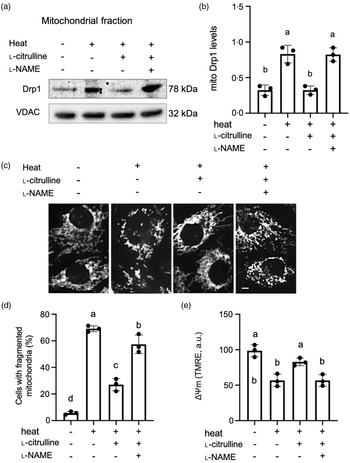
The literature has reported that heat stress may increase NO levels in plants and animals, which depends on the types of tissue or cells, severity and duration of heat treatment(Reference Parankusam, Adimulam and Bhatnagar-Mathur30,Reference Gerstberger31) . In our study, NO levels did not increase in C2C12 myoblasts exposed to 43°C heat for 2 h. Similar to data collected at the normal 37oC temperature, l-citrulline increased NO levels under heat stress at 43°C. The increase in NO was also blocked by l-NAME (online Supplementary Fig. 1). Upon heat exposure, we discovered an increased localization of Drp1 to mitochondria (Fig. 5(a) and (b)). Mitochondria became short and fragmented (Fig. 5(c)), and the percentage of cells containing fragmented mitochondria increased from 6.9 % in normal temperature cells to 61.9 % in heat-exposed cells (P < 0.01, Fig. 5(d)). The ΔΨm was also significantly decreased in myoblasts exposed to heat (Fig. 5(e)). Notably, l-citrulline treatment prevented heat-induced translocation of Drp1 to mitochondria (Fig. 5(a) and (b)), maintained tubular mitochondrial morphology and preserved ΔΨm (Fig. 5(c)–(e)). l-NAME abolished all of the above protective effects by l-citrulline under heat stress (Fig. 5(a)–(e)).
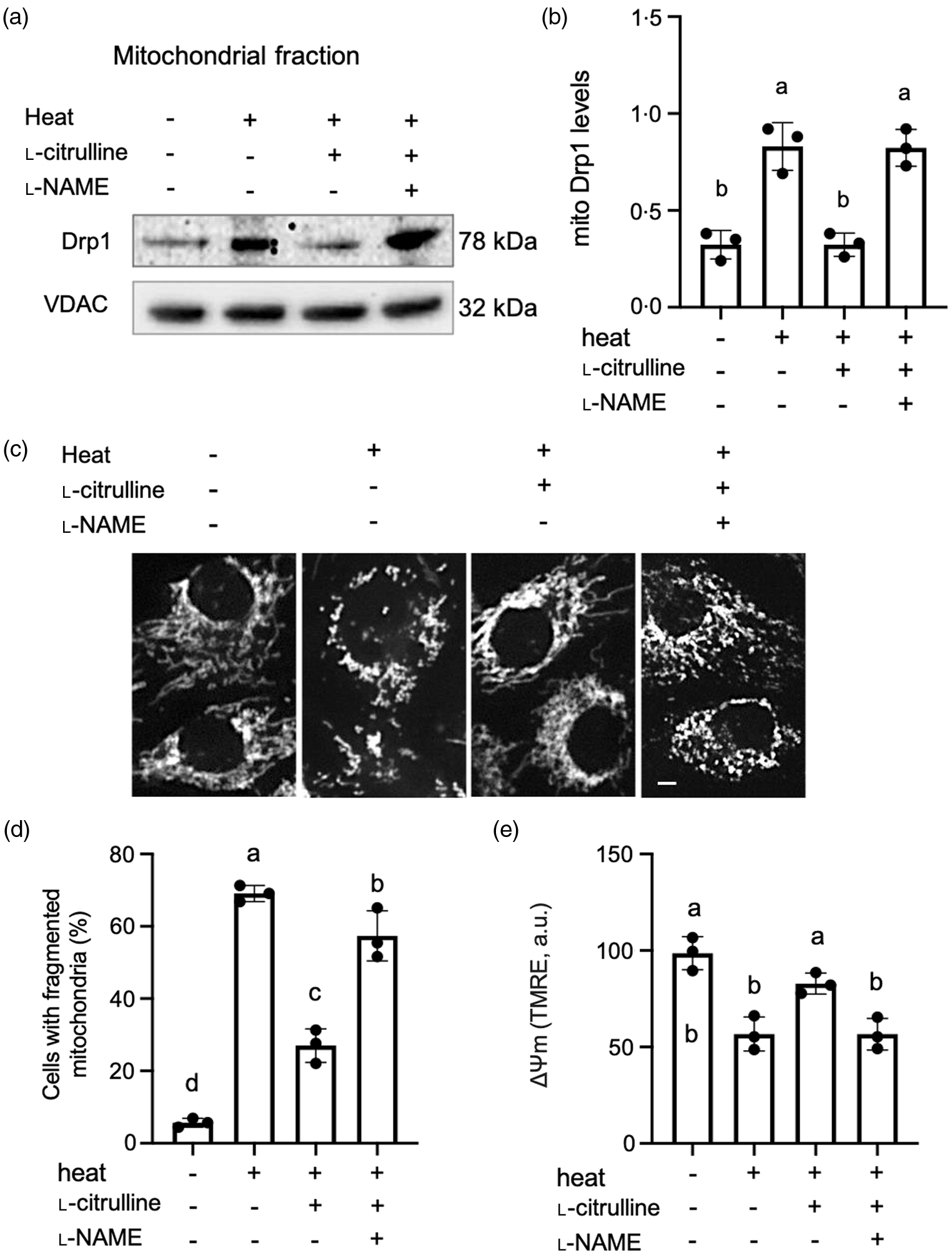
Fig. 5. l-Citrulline prevents heat-induced translocation of Drp1 to mitochondria, mitochondrial fragmentation and decreased ΔΨm. (a) Representative western blot images of Drp1 in the mitochondrial fractions, VDAC served as a mitochondrial loading control. (b) Densitometric analysis of mitochondrial (mito) Drp1 levels (ratio to VDAC). (c) Representative fluorescent microscopy images of C2C12 myoblasts with mitochondria labelled by MitoTracker Red to determine mitochondrial morphology. Scale bar: 5 µm. (d) Percentage of myoblasts with fragmented mitochondria, 200–300 cells were randomly counted for each condition. (e) ΔΨm was determined by TMRE fluorescence intensity (a.u.), 30 cells were randomly selected for each condition. Cells were pretreated with phosphate-buffered saline (control), 2 mM l-citrulline and 1 mM l-NAME, then incubated at 37°C or 43°C heat for 2 h. Values are means ± sd; n = 3 independent experiments. Labelled means without a common letter differ, P < 0.05.
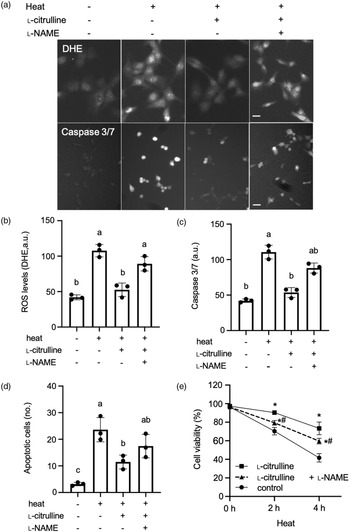
l-Citrulline prevents heat-induced cellular reactive oxygen species production and cell injury in C2C12 myoblasts
We finally tested whether l-citrulline treatment could block heat-induced oxidative injury in C2C12 myoblasts. Heat exposure led to a 265 % increase in cellular ROS levels (P < 0.05), which was blocked by l-citrulline treatment (Fig. 6(a) and (b)). In addition, l-citrulline inhibited caspase 3/7 activation (Fig. 6(a) and (c)) and apoptotic cell death (Fig. 6(d)) and improved cell viability (Fig. 6(e)) under heat stress conditions. Furthermore, inhibition of NO by l-NAME under heat stress abolished the protective effects of l-citrulline on ROS production, caspase activation, apoptosis and cell viability (Fig. 6(a)–(f)).

Fig. 6. l-Citrulline prevents heat-induced cellular reactive oxygen species (ROS) production and cell injury. (a) Representative fluorescent microscopy images of C2C12 myoblasts labelled with ROS indicator dihydroethidium (top) and caspase-3/7 green detection reagent (bottom). Quantification of ROS levels (b) and caspase-3/7 activities (c). Thirty cells were randomly selected for quantification in each group. (d) Numbers (no.) of annexin V positive (apoptotic) cells, and (e) Cell viability. Cells were treated with phosphate-buffered saline (control) or 2 mM l-citrulline, then exposed to 37°C or 43°C for 2 h or indicated times; l-NAME (1 mM) is a non-selective nitric oxide synthase inhibitor. Scale bars: 10 µm. Values are means ± sd; n = 3 independent experiments. Labelled means without a common letter differ, P < 0.05. *P < 0.05 v. control and # P < 0.05 v. l-citrulline.
Discussion
The overall morphology of mitochondrial networks is determined by the balance between mitochondrial fission and fusion(Reference Yu, Wang and Yoon5). Our previous studies have shown that heat exposure causes excessive mitochondrial fragmentation and dysfunction, which contribute to heat-induced ROS production and skeletal muscle injury(Reference Yu, Ferdjallah and Elenberg11,Reference Yu, Deuster and Chen12) . The goal of this research was to explore the downstream effects of l-citrulline, specifically on l-citrulline-induced NO production on mitochondrial morphology and function in myocytes, and to test whether l-citrulline could protect against heat-induced oxidative injury. Here, we report three novel findings. First, we find that l-citrulline increases NO production, which then suppresses Drp1-mediated mitochondrial fission. Second, l-citrulline preserves mitochondrial morphology and function under heat stress conditions. Third, by preserving mitochondrial morphology and function, l-citrulline reduces the impact of heat-induced increase in ROS and cell injury. These data provide a mechanism whereby l-citrulline shifts the balance of mitochondrial dynamics toward fusion in myocytes and affords protection during periods of heat stress.
Consistent with previous findings(Reference Yu, Ferdjallah and Elenberg11,Reference Yu, Deuster and Chen12,Reference White, Saleh and Nonner32) , we found that heat exposure causes the ΔΨm collapse in myocytes. Mitochondria coordinate cell-wide stress responses and determine cell life-or-death(Reference Yu, Wang and Yoon5,Reference Manoli, Alesci and Blackman33,Reference Suliman and Piantadosi34) . As the powerhouse of the cell, mitochondrial electron transport chain moves protons across the inner membrane to generate the ΔΨm that provides the driving force for ATP synthesis(Reference Yu, Wang and Yoon5). Mitochondria are not only the main cellular source but also a major target of ROS, and serve a critical role in regulating apoptosis(Reference Yu, Wang and Yoon5,Reference Youle and van der Bliek35,Reference Hoppins and Nunnari36) . In response to stress, that is, heat in this study, mitochondria produce more ATP to meet the growing energy demands for survival. They also increase the generation of reactive metabolic intermediates and ROS as irreversible byproducts during electron flow. Such processes result in the accumulation of ROS within the mitochondria and opening of the mitochondrial permeability transition pore, subsequently causing a reduction in ΔΨm.
A mild depolarisation of mitochondria under physiological conditions may still be potent enough to make ATP, but leak less electrons to oxygen to produce superoxide: this represents an antioxidant defense mechanism for the balance of redox hemostasis(Reference Vyssokikh, Holtze and Averina37,Reference Wang, Gong and Wang38) . Loss of ΔΨm within physiological ranges is transient (recover in a few seconds), likely occur in only a subset of individual mitochondria, and requires an intact mitochondrial electron transport chain(Reference Wang, Gong and Wang38). Whereas our current and previous studies show that heat-induced collapses in ΔΨm are extended and can be detected in whole-cell mitochondria, the collapse is often accompanied by excessive fragmentation of mitochondria and disorganised cristae structure, with less electron dense and swollen configurations(Reference Yu, Ferdjallah and Elenberg11,Reference Yu, Deuster and Chen12) . All of these indicate mitochondrial dysfunction under heat stress, which is associated with increased production of ROS, release of cytochrome c into the cytosol and subsequent caspase activation and cell death(Reference Yu, Ferdjallah and Elenberg11,Reference Yu, Deuster and Chen12,Reference Slimen, Najar and Ghram39,Reference Wang, Cai and Chen40) . Mitochondrial dysfunction contributes to the pathogenesis of heat-related illness.
As dynamic organelles, mitochondria constantly change shape through frequent fission and fusion(Reference Yu, Wang and Yoon5,Reference Yoon, Galloway and Jhun6) . Proper control of mitochondrial morphology is critical for normal cell function, as defects in proteins involved in mitochondrial fission and fusion can cause human diseases(Reference Chan41,Reference Archer42) . Several studies have shown that heat stress activates the mitochondrial fission protein Drp1 and degrades the fusion protein OPA1(Reference Yu, Ferdjallah and Elenberg11–Reference Sanjuan Szklarz and Scorrano13). Excessive mitochondrial fragmentation may disrupt the structural organisation of the electron transport chain on the inner mitochondrial membranes and impair its function(Reference Yu, Wang and Yoon5,Reference Yoon, Galloway and Jhun6) . Importantly, inhibiting mitochondrial fission not only maintained normal mitochondrial structure and function under heat stress, but also protected against heat-induced ROS production and skeletal muscle injury(Reference Yu, Ferdjallah and Elenberg11,Reference Yu, Deuster and Chen12) . Therefore, Drp1-mediated mitochondrial fission may serve as a target for preventing heat-related injury.
l-Citrulline is a non-essential amino acid that is not only found in food; the body can also synthesise it from l-glutamine and l-arginine by intestinal enterocytes. Although it is controversial whether glutamine is a precursor for citrulline synthesis, supplemental glutamine can increase citrulline synthesis in animals and humans(Reference Marini43). Our recent study shows that glutamine is essential for mitochondrial function, and glutamine depletion impairs the heat-shock response in C2C12 myoblasts(Reference Dohl, Passos and Foldi44). l-Citrulline levels are decreased in chickens exposed to heat(Reference Chowdhury, Tomonaga and Ikegami22), and l-citrulline supplementation improves physiological performance in chicken and swine under heat stress conditions(Reference Uyanga, Wang and Tong17,Reference Liu, de Ruyter and Athorn45) . The present study offers more evidence for a protective effect of l-citrulline against heat stress: l-citrulline ameliorates heat-induced ROS production and cell injury in mouse C2C12 myoblasts.
Our results show that l-citrulline treatment increases NO levels in C2C12 myoblasts, and this increase is abolished by the NO synthase inhibitor l-NAME. NO is a gaseous signalling molecule that regulates many physiological processes, including mitochondrial metabolism(Reference Poderoso, Helfenberger and Poderoso46). Excessive NO stimulates S-nitrosylation of Drp1 in neurons, which promotes mitochondrial fission and contributes to the pathogenesis of Alzheimer’s disease(Reference Cho, Nakamura and Fang21). Another study reported conflicting results – that S-nitrosylation of Drp1 has no influence on fission activity and is not specific to Alzheimer’s disease(Reference Bossy, Petrilli and Klinglmayr47). Lee and Kim recently suggested that S-nitrosylation of Drp1 regulates its phosphorylation and fission activity in CA1 neurons(Reference Lee and Kim48). Our results show that l-citrulline increases NO levels, which in turn inhibits fission via regulating Drp1 phosphorylation, but not S-nitrosylation in C2C12 myoblasts. Likely, the effect of NO on mitochondria fission and its regulatory mechanism is dependent on many factors, such as cell types, stress and/or NO levels.
Drp1 belongs to the dynamin family of large GTPases and primarily localises in the cytosol. Upon activation, it is recruited to the mitochondrial outer membrane, where it assembles into a ring that encircles mitochondria and drives division via GTP-dependent constriction(Reference Yu, Wang and Yoon5,Reference Chan41) . Phosphorylation of Drp1 represents an important regulatory mechanism for fission and several phosphorylation sites have been characterised. It has been shown that phosphorylation of Drp1 at Ser 616 activates mitochondrial fission, whereas phosphorylation of Drp1 at Ser 637 decreases its GTPase activity and inhibits fission(Reference Chang and Blackstone49–Reference Cereghetti, Stangherlin and Martins de Brito51). In this study, l-citrulline-induced NO production decreased Drp1 protein expression and Ser 616 phosphorylation, and increased Ser 637 phosphorylation, which resulted in a reduction in Drp1 levels in the mitochondria. The proteins involved in mitochondrial fusion (Mfn1, Mfn2 and OPA1) remained unchanged, thereby leading to mitochondrial elongation in l-citrulline-treated cells. The cGMP-dependent protein kinase may serve as a signalling pathway that mediates Drp1 phosphorylation by NO(Reference De Palma, Falcone and Pisoni52).
Under physiological conditions, mitochondrial fission facilitates the segregation of damaged mitochondria into autophagosomes for subsequent degradation by mitophagy, which is essential for maintaining a healthy mitochondrial population and cell fitness(Reference Twig, Elorza and Molina53). Our recent study shows that severe heat exposure promotes mitochondrial fission and induces mitophagy in mouse hypothalamus(Reference Chen, Yu and Deuster54). Interestingly, repeat exposure of C2C12 cells or mice to mild heat (heat acclimation) promotes fusion and evokes a stress response named stress-induced mitochondrial hyperfusion(Reference Yu, Deuster and Chen12), a known pro-survival response that protects mitochondria from autophagic removal when they are most needed to fight back against stress(Reference Youle and van der Bliek35). In addition to the above-discussed mitochondrial quality control mechanism by regulating mitophagy, mitochondrial fission has multiple essential functions in cells, including the appropriate distribution of mitochondria to daughter cells during mitosis(Reference Kashatus, Lim and Brady55) and serving a critical role in myogenic differentiation(Reference De Palma, Falcone and Pisoni52,Reference Kim, Kim and Yoon56) . The present study finds that l-citrulline induces mitochondrial elongation and prevents cell injury under acute heat stress; the effect of long-term l-citrulline supplementation on fission inhibition and cell function remains to be further investigated. Several studies have shown that l-citrulline supplementation over an extended period of time has no disadvantages in cell growth and muscle differentiation(Reference Ham, Gleeson and Chee15,Reference Vadivel, Aschner and Rey-Parra26) . Likely, whereas l-citrulline helps maximise oxidative capacity under stressful conditions, cells are still capable of maintaining the rates of mitochondrial fission and fusion to meet their needs and function properly.
In summary, it has been proposed that elongated mitochondria are more efficient for ATP production(Reference Yu, Wang and Yoon5), preserve mitochondrial contents by segregating them from autophagic removal(Reference Youle and van der Bliek35,Reference Twig, Elorza and Molina53) and enhance cell survival and prevent cell death under stress conditions(Reference Youle and van der Bliek35). Mitochondrial fission and dysfunction are commonly documented in a number of pathological conditions such as diabetes, neurodegeneration, CVD and heat injury(Reference Yoon, Galloway and Jhun6,Reference Yu, Ferdjallah and Elenberg11,Reference Yu, Deuster and Chen12,Reference Chan41,Reference Archer42,Reference Wang, Yu and Lee57) . As discussed above, l-citrulline increases NO levels and shifts mitochondrial dynamics balance favouring mitochondrial fusion/elongation, thus preventing mitochondrial fragmentation and cell injury during heat stress (online Supplementary Fig. 2). l-Citrulline may be an effective treatment choice for heat-related illnesses and other disorders that result from a functional failure of the mitochondria.
Acknowledgements
We thank Dr. Beth McNally for critical reading of the manuscript. This study was supported by US Department of Defense (DoD) the Defense Health Program (HU00011920047).
The opinions and assertions expressed herein are those of the author(s) and do not reflect the official policy or position of the Uniformed Services University or the Department of Defense. The contents of this publication are the sole responsibility of the author(s) and do not necessarily reflect the views, opinions or policies of The Henry M. Jackson Foundation for the Advancement of Military Medicine, Inc. Mention of trade names, commercial products or organisations does not imply endorsement by the US Government. The authors have no financial interests or relationships to disclose.
T. Y., L. W. and P. D. conceived study; T. Y. designed experiments; T. Y., Y. P. and L. W. conducted research; T. Y. analysed data and wrote the paper; T. Y., Y. P. L. W. and P. D. edited the paper. All authors read and approved the final manuscript.
Supplementary material
For supplementary material/s referred to in this article, please visit https://doi.org/10.1017/S0007114522001982