



Introduction
Nerve conduction studies (NCS) and electromyography (EMG) are electrodiagnostic studies that assist with confirming and localizing lesions within the peripheral nervous system. NCS involves delivering small electrical stimuli over peripheral sensory, motor, or mixed nerves and measuring the responses that are obtained. Reference Mallik and Weir1 EMG in children utilizes a small needle electrode that records electrical activity within a muscle of interest, providing auditory and visual information from a resting muscle as well as motor unit action potentials (MUAPs) that are generated during muscle contraction. Together, NCS/EMG can identify features of a sensory or motor-predominant neuropathy, polyneuropathy with demyelinating and/or axonal features, primary disorder of muscle or a disorder of neuromuscular transmission.
The neurological examination may be limited or unreliable in young or developmentally delayed children. As such, NCS/EMG may be particularly helpful at providing information pertaining to sensory nerve involvement that may not be assessed on clinical examination alone.
NCS/EMG results can help direct genetic testing, for example, ensuring the most appropriate gene panel is pursued in a child with a suspected neuromuscular disease. Even for children with complex inherited neurological diseases where dystonia, spasticity, ataxia, or cognitive impairment may be the primary presenting feature, NCS/EMG can allow for improved phenotyping if a concomitant neuropathy is detected, which has the potential to narrow the differential diagnosis considerably. Reference Rossor2 Improved access to genetic testing has obviated NCS/EMG for some diseases such as spinal muscular atrophy (SMA) or Duchenne muscular dystrophy (DMD), which has been postulated to be one of the reasons why there has been a trend for fewer infants or younger children to receive NCS/EMG at some centers. Reference Karakis, Liew, Darras, Jones and Kang3 Nevertheless, NCS/EMG remains a valuable tool for children with ultra-rare genetic disorders as well as the interpretation of variants of uncertain significance (VUS) in exome data. NCS/EMG also remains an important tool for the diagnosis of acquired diseases of peripheral nerve, muscle, or neuromuscular junctions that have the potential to mimic hereditary conditions. Prior studies have reported 30–50% of peripheral neuropathies in children have been estimated to arise from acquired causes, emphasizing the need for careful clinical and NCS/EMG testing to yield a diagnosis that would not be available from genetic testing alone. Reference Shabo, Pasman, van Alfen and Willemsen4-Reference Evans6
Our goal was to review the volume and referral sources of NCS/EMG at our center as well as to understand the diagnostic utility and tolerability of NCS/EMG performed at our pediatric tertiary care hospital.
Methods
This study involved a retrospective chart review approved by the local Research Ethics Board (#19/139X). Inclusion criteria included all pediatric patients (0–18 years old) who had a NCS/EMG study performed at the Children’s Hospital of Eastern Ontario from 2014 to 2019. Children who had a NCS/EMG performed at a different institution or were seen purely for a clinical consultation (without neurophysiological testing) were excluded. A list of patients seen during the above time period was obtained from hospital records. This was cross-referenced with the billing code for NCS/EMG during the same time to ensure that all patients were captured. The data collection instrument was a REDCap database, Reference Harris, Taylor, Thielke, Payne, Gonzalez and Conde7 with the following non-identifiable variables extracted from the medical chart of patients: age of patient, referring physician’s specialty, indications for test, if the test was performed awake or under sedation, the electrophysiological findings status (normal/abnormal), site, and final clinical diagnosis. The entire available electronic patient chart, including reports available beyond the study date, was reviewed to determine the final clinical diagnosis.
All statistical analyses were carried out using R version (4.0.3). 8 Descriptive statistics were calculated using mean and standard deviation (SD) or median and interquartile range (IQR) for continuous variables, and frequency and percentage for categorical variables. The overall monthly trend over time was presented graphically and we tested for a difference in this trend over time using a quasi-Poisson regression adjusted for autocorrelation. We also tested if there is a change in seasonality by conducting a Box–Pierce test. To test the trend in the proportion of yearly studies referred by different types of physicians, we conducted the Cochran–Armitage test for trend. Reference Armitage9 Percentages were accompanied by 95% confidence intervals (CI). Two-sided p-values less than 0.05 were considered statistically significant.
Results
Patient and Study Characteristics
From 2014 to 2019, 725 NCS/EMG studies were performed at our hospital. Data were available for all of 2015 to 2019 but only for part of 2014 (from October to December). The number of studies performed each year were as follows: 2019 (122 studies); 2018 (149); 2017 (120); 2016 (158); 2015 (147), and 2014 (29). The median age of patients was 13.2 years old (IQR 7.9, 15.9 years) with an equal division of studies performed by gender (Table 1). NCS/EMG was performed most commonly in patients from 14 to 17 years old (Figure 1). Most studies were performed on an outpatient basis (617; 85.2%), at either an ambulatory care setting or, in the case of sedated studies, at the surgical day unit. Inpatient studies were performed for 107 (14.8%) patients who were admitted to hospital.
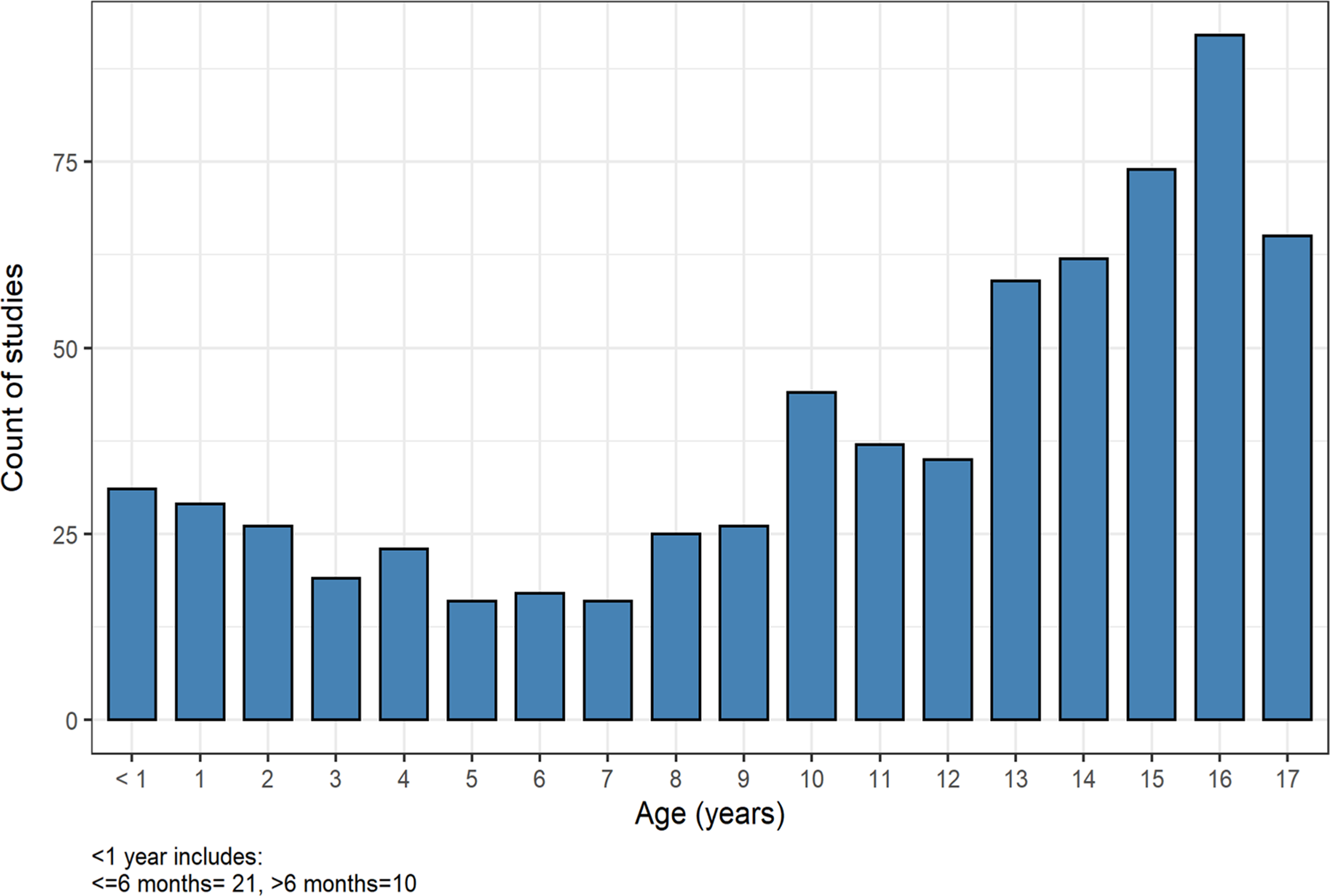
Figure 1: Age of patients undergoing NCS/EMG study (2015–2019). Legend: Number of studies performed for patient’s age from 2015 to 2019 showing a higher number of studies performed in older patients (14–17 years).
Table 1: Patient and study characteristics
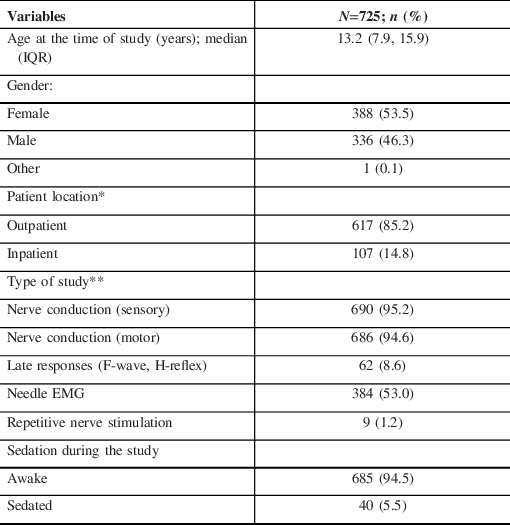
*Outpatient location refers to patients seen in an ambulatory care clinic or in the day surgery unit (for sedated studies); the location of one (1) patient was not specified.
**Percentages for type of study to not add up to 100% as patients may have had more than one study.
Most studies included NCS (690/725; 95%), while over half of studies included an EMG (384; 53%). Less commonly performed were late responses (defined as H reflex and F waves 62; 8.6%), repetitive nerve stimulation (RNS) testing (9; 1.2%), or blink responses (6; 0.8%), when clinically indicated.
Follow-up studies accounted for 16.1% of studies (117/725). Of these, 53/117 (45.3%) were follow-up of acquired neuromuscular conditions, most commonly a traumatic mononeuropathy or plexopathy, a tumor-related mononeuropathy (perineurioma plexiform neurofibroma), or less commonly, an inflammatory polyneuropathy (ie, GBS [Guillain-Barré syndrome] or CIDP [chronic inflammatory demyelinating polyneuropathy]). Interestingly, 37.6% (44/117) of follow-up studies were normal. On these occasions, studies were most commonly performed to investigate recurrent or persistent sensory symptoms despite previously normal studies. Many were ultimately found to have an orthopedic cause (myofascial pain syndrome) or a psychiatric cause (conversion disorder). A subset of repeat studies were performed for genetic/metabolic disorders that can be associated with a progressive compressive neuropathy (mucopolysaccharidosis) or a progressive polyneuropathy (cobalamin C deficiency and mitochondrial disorder). We report 13% (15/117) follow-up studies were performed in patients with a hereditary neuromuscular disorder and 6% (7/117) were required in patients with yet-unidentified progressive neurological disorders.
Number of Studies Over Time
There was no change in the monthly rate of total number of studies over the time (Estimate = −0.001; 95% CI= −0.006, 0.005, p = 0.76). In this data pooled across referral sources, the need for NCS/EMG studies did not statistically change during this 5-year period. When stratified by age group, and comparing yearly rates, only the (1–5 years) age group showed a decrease in NCS/EMG studies over time (p = 0.004, Bonferroni corrected). Caution is needed interpreting this evidence as it is based on a time series of only 5 years.
Referral Patterns
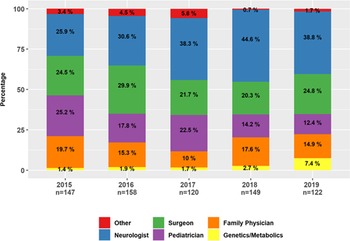
Referral rates of NCS/EMG studies were evaluated for the 5 years that complete data sets were available (2015–2019) with N = 693 studies performed during that time (Figure 2). Referrals for NCS/EMG were from other neurologists (35.4%; 245/693), surgeons (24.4%; 169/693), pediatricians (18.5%; 128/693), family physicians (15.7%; 109/693), genetics or metabolics (2.9%; 20/693) or other (3.2 % 22/693). Referrals from surgery were from orthopedic (58%), plastic surgery (36%), or neurosurgery (6%). Family medicine physicians with additional training in sports medicine were all grouped as family physicians.

Figure 2: Trend in referral source of pediatric NCS/EMG (2015–2019). Legend: Stacked barplot showing the proportion of specialists making referrals for NCS/EMG studies, by year.
Several trends were observed over the 5-year period. While the overall number of NCS/EMG studies was constant, the proportion of referrals from genetics and metabolic specialists increased from 1.4% to 7.4% (p < 0.05). Similarly, the proportion of referrals from other neurologists increased from 25.9% to 38.8%. Referrals from pediatricians showed the greatest proportional decline from 25.2% to 12.4% (p < 0.05). No significant change was noted in the proportion of referrals from family physicians or surgeons (Figure 2). Of the NCS/EMG requests from genetics/metabolic specialists, 20% (4/21) were requested to assist with the interpretation of VUS. Another 50% (10/21) were referred to assist with phenotyping of patients with a yet-undiagnosed neurological disorder. Thirty percent (7/21) were referred for NCS/EMG to assist with phenotyping or provide surveillance for a child with a known genetic or metabolic disorder associated with compressive neuropathies or polyneuropathy.
Tolerability
NCS/EMG were largely performed on awake, non-sedated patients (94.5%; 685/725) with a smaller proportion of patients requiring general anesthetic (6.5%; 40/725).
For most patients (95.2%; 690/725), a complete study was performed, meaning that the neurophysiological information was able to meet the goals of the electromyographer and answer the clinical question that had been posed. In a smaller subset of patients, the NCS/EMG was terminated prematurely (4.8%; 35/725). Reasons for premature termination of the study included anxiety (45.7%; 16/35), patient pain, refusal, and/or difficulty tolerating the procedure (40%; 14/35) as well as one each (2.9%; 1/35) of the following: bandages covering the part of the area to be studied (1 patient); anticoagulation (limiting EMG; 1 patient); electrical interference that could not be eliminated (1 patient); time limitations given other sedated procedures required (1 patient); and reason not specified (1 patient). Of note, studies that were terminated prematurely may still have provided some diagnostic information. For example, a patient with a family history of Charcot–Marie–Tooth (CMT) disease may have only been able to tolerate a limited NCS studying a median nerve sensory and motor response. Although a lower limb may not have been studied, if conduction velocity was slow (<75% below lower limit of normal for age), this may have provided clinically useful information. Studies that were sedated or prematurely terminated tended to occur more frequently in patients with age groups <10 years old compared to those >10 years old (Figure 3).
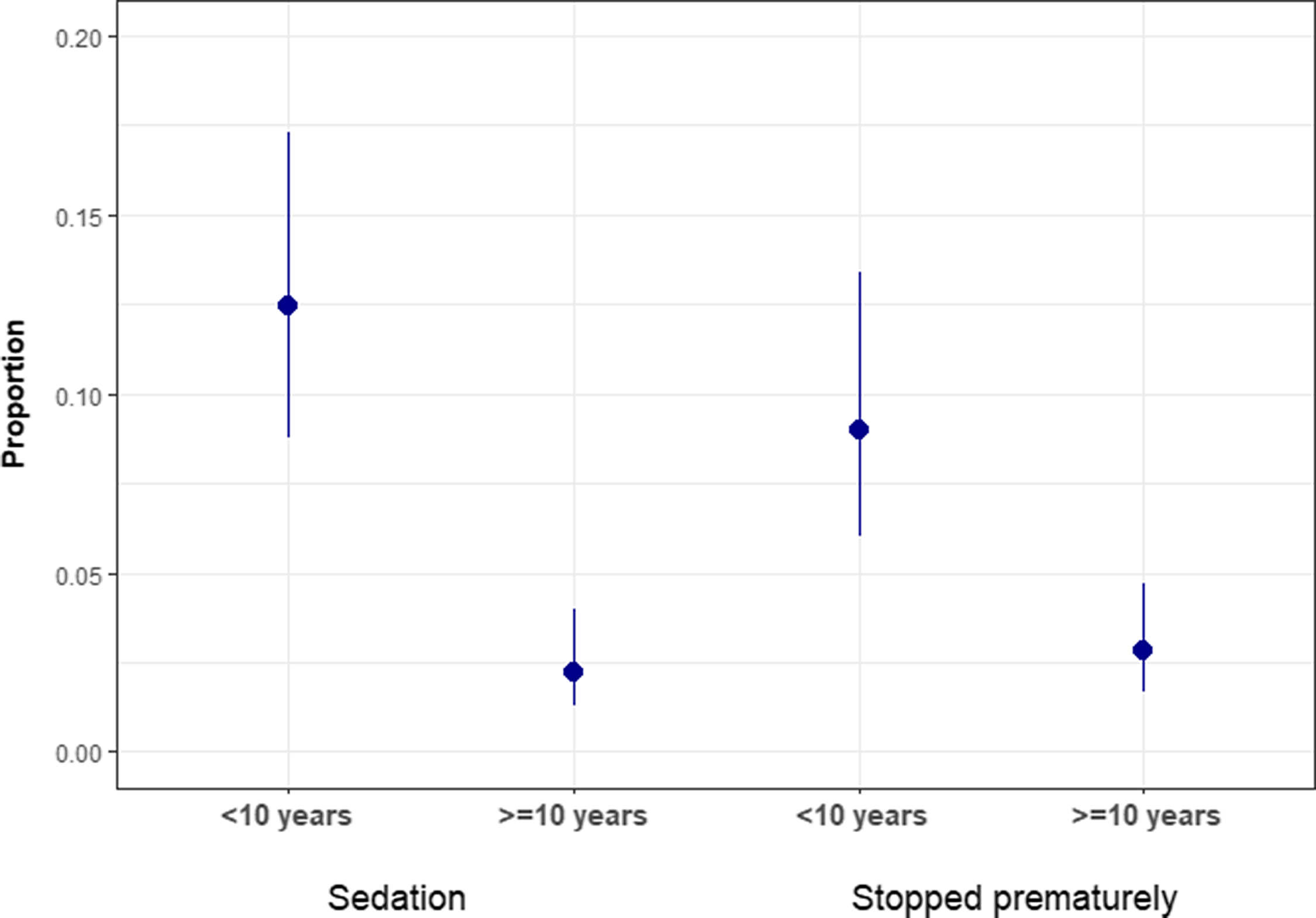
Figure 3: Proportion of studies performed under sedation or stopped prematurely by age group. Legend: Proportion of studies performed under sedation or were stopped prematurely by age of the patient (<10 years old vs. >10 years old). Error bar with 95% CI showing the proportion of pediatric patients referred for NCS/EMG studies that had sedations and their study stopped prematurely.
There were 40 sedated studies. All (100%) included NCS and most (80%; 32/40) included EMG. These sedated studies were often coordinated with magnetic resonance imaging (MRI) and in such cases, performed immediately after MRI while the patient was still sedated. On occasions where an EMG was not included, it was either because it was not ordered by the referring physician and/or the neurologist/electromyographer was not available at the time of the MRI. Interestingly, there were no RNS (repetitive nerve stimulation) or SSFEMG (stimulated single fiber EMG) performed under sedation during the study period 2014–2019.
Diagnostic Utility
Neurophysiological results were available for 725 studies. Of these, 4.1% were reported to be nondiagnostic, either because the study was terminated prematurely (2.2%; 16/725) or while abnormalities were noted it was non-localizable and as such was a nondiagnostic study (1.9%; 14/725).
NCS/EMG reported abnormalities in 44.1% (320/725) of patients, while 55.9% (405/725) showed no neurophysiological abnormalities. The most common abnormalities reported on NCS/EMG included mononeuropathy (18.2%; 132/725), polyneuropathy (12.4%; 90/725), myopathy (4.1%; 30/725), motor neuronopathy or neuropathy (4.0%; 29/725), plexopathy (3.4%; 25/725), radiculopathy (1.4%; 10/725), and/or a disorder of neuromuscular transmission (0.1%; 1/725).
For patients who had a normal NCS/EMG (55.9%), the final clinical diagnosis included disorders isolated to the central nervous system (11.4%), somatization or conversion disorder (3.4%), or other non-neurological disorders including, as examples, myofascial pain or tendonitis.
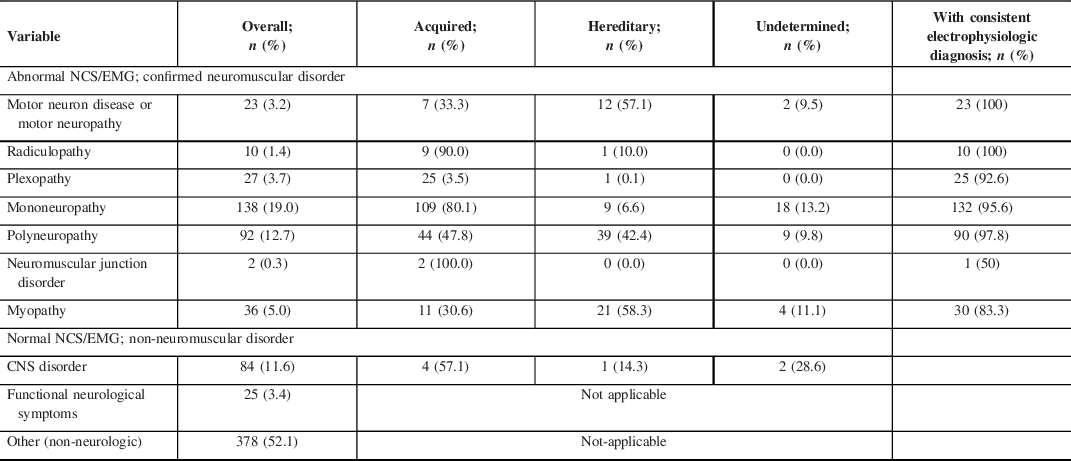
For patients with confirmed neuromuscular abnormalities (45.2%; 328/725), most were acquired disorders (63.1%; 207/328) such as traumatic mononeuropathy or plexopathy. By comparison, 25.9% (85/328) were hereditary and 11.0% undetermined. Mononeuropathies were the most likely to have a specified etiology with 80.1% due to an acquired cause (typically traumatic), and polyneuropathies were more evenly divided with 47.8% due to acquired and 42.4% due to a hereditary cause (Table 2).
Table 2: Electrophysiologic and clinical diagnoses of 725 patients, across acquired and hereditary diseases
Discussion
NCS/EMG remains an important tool for evaluation of children with a suspected neuromuscular disease. Despite advances in accessibility and affordability of genetic testing, this diagnostic tool remains valuable. Reference Rossor2 The number of NCS/EMG studies remained relatively constant at our center during this 5-year window when neuromuscular gene panels were clinically accessible and exome sequencing was performed with greater frequency.
Regarding the types of studies conducted, we want to discuss a couple points. While almost all studies included NCS, just over half included EMG. We interpret the inclusion of EMG in 53% (384/725) of all studies as a consequence of two main factors, as studies with NCS alone were more frequently requested by family physician, neurologists, or geneticists.
First, neurophysiological studies are often requested by external referral to our center. Referring physicians were given the option of: (1) NCS alone or (2) NCS/EMG plus neuromuscular consult. If a referring physician (often a family physician or a pediatrician) requested a NCS alone, the technologist would perform the electrodiagnostic test according to their assessment. The technologist may or may not consult with the neurologist in advance to ensure that the appropriate nerves were studied. Although the neurologist would not be present for this test, it would be stated clearly in the report if they felt that an EMG with neuromuscular consult was clinically indicated.
Second, while we recognize that EMG is a very important element of the neurophysiological testing, as it is both more sensitive than NCS at identifying early neuropathy and is uniquely able to identify myopathic changes, it can be difficult for some children to tolerate. In some conditions (e.g., testing a child with a family history of CMT type 1, evaluating an ataxic child with possible Friedrich ataxia to look for sensory nerve involvement and/or evaluating for possible carpal tunnel syndrome), EMG may have a lesser role and in our opinion, can be safely omitted. As neurologists and geneticists tended to request more of the NCS-only studies, it is likely that they were confident their specific question could be answered without EMG.
There were very few neuromuscular junction studies during the study period, with only nine non-sedated RNS studies performed. We attribute this to the very small number of children with neuromuscular junction disorders that presented during 2014–2019. We confirmed with medical records, for example, that there only three children diagnosed with new-onset juvenile myasthenia gravis from 2015 to 2019, compared to eight children diagnosed with new-onset JMG from 2010 to 2014. Infants and children with possible congenital myasthenic syndrome tending to undergo gene panels and/or exome sequencing as a first-line investigation which has often obviated the need for a sedated SSFEMG.
Regarding referral patterns, it is noteworthy that we did observe evidence of an increase in the requests for NCS/EMG from genetic and metabolic subspecialists. As noted above, some of these referrals (4/21) were to assist in interpreting a variant of unknown significance, half (10/21) were to assist in phenotyping a patient with yet-undiagnosed presumed genetic syndrome, and the remaining 7/21 were for monitoring of patients with diagnosed genetic syndromes known to be associated with neuropathies. The increase in the proportion of referrals from neurologists could similarly be related to that, as well as to an increased awareness that even for children with a complex neurological disease where neuromuscular features may not be the primary or presenting feature, NCS/EMG has the potential to improve phenotyping and narrow the differential diagnosis if abnormal findings are observed. Reference Rossor2 The significant reduction in the proportion of referrals from pediatricians could reflect a shift in practice towards the appropriate request of genetic testing for the more common neuromuscular disorders (i.e., SMA, myotonic dystrophy, DMD). These findings are aligned with a recent consensus statement from the American Association of Neuromuscular and Electrodiagnostic Medicine (AANEM) which also reported changing referral patterns, with fewer referrals for genetic conditions such SMA, but a compensatory increase in referrals for acquired conditions including trauma which overall gives rise to stable or even increasing NCS/EMG referral numbers. Reference Kang, McMillan and Kuntz10
For children with acquired neuromuscular disorders, NCS/EMG remains a particularly valuable diagnostic tool. Acquired neuromuscular disorders can arise from a broad range of causes with categories, including trauma, infectious, post-infectious or autoimmune, toxic exposure, or neoplastic. NCS/EMG is less commonly used for some conditions such as juvenile dermatomyositis, Reference Kobayashi, Akioka and Kobayashi11 while it remains an important diagnostic criteria for others such as CIDP. Reference Nevo and Topaloglu12
Overall, acquired neuromuscular diseases also comprise a significant proportion of neuromuscular diseases with 30–50% of childhood peripheral neuropathies attributed to acquired disorders. Reference Ouvrier and McLeod5,Reference Evans6 Although these two studies are now about 30 years old, the relative proportion acquired studies is similar to that obtained in our current sample where 63.1% of children with confirmed neuromuscular disorders were found to have an acquired cause, most commonly from a prior trauma. Although it is appropriate for NCS/EMG to be delayed several weeks after a traumatic injury, a properly timed study can provide useful information not only to localize the site of the lesion but also to confirm if axonal continuity is or is not present. In infants who have sustained a neonatal brachial plexus palsy, the absence of MUAPs in the biceps at 3 months old is one of the proposed criteria that would favor surgical intervention. Reference Wilson, Chang and Yang13 The value of NCS/EMG at our medium-sized Canadian pediatric hospital is congruent with a report from a Turkish hospital which performed approximately 578 pediatric NCS/EMG studies per year. Reference Kocasoy Orhan, Baysal Kirac and Yalinay Dikmen14
Our observation that the majority of Canadian children (95.2%) were able to tolerate a non-sedated NCS/EMG study is also important when counselling families. This was true even for the younger children (< 10 years old; Figure 3), although this age group had slightly higher rates of sedated or prematurely terminated studies.
There are some strengths and limitations associated with our retrospective study. The strengths of this study include confidence that all NCS/EMG studies were captured during the above time window, since this procedure is associated with a unique billing code in our province making it easy to ensure all studies were captured. The authors ensured that data collection was performed by a trained Research Coordinator (JVG) and a Pediatric Neurology Resident with Canadian EMG Board certification (KMJ) to minimize the risk of error. Data were collected from the unique NCS/EMG clinical reports that are generated for every patient who has this procedure performed at our center. Nevertheless, since the data were collected retrospectively, there is the potential for error in final diagnosis which is an inherent limitation of all retrospective studies. For example, if a patient was to have a NCS/EMG performed and then move to a different city, the final diagnosis may not be known. Another limitation is that diagnostic performance metrics were not included as objective in this study, and thus we cannot provide information regarding false-negative or false-positive rates. Future studies are required to provide better estimates of the diagnostic accuracy of NCS/EMG.
Overall, our study confirms that NCS/EMG in children are largely well tolerated and provide clinically important information even with the tremendous advances seen in genetic testing.
Acknowledgements
The authors want to thank Dr Jemila Hamid for her contribution in the analysis plan for this study and Sjjad Dehnoei for his assistance in the statistical trend analysis.
Disclosures
No authors report any disclosures relevant to the manuscript.
Statement of Authorship
KMJ designed and conceptualized the study, participated in data collection, and drafted the manuscript. JM participated in data collection. RW and LH provided data analysis. HJM contributed to the design and conceptualization of the study and assisted with writing of the manuscript. All authors provided critical review and edited the manuscript. All authors approve of the manuscript in its current form.





 Open access
Open access