



Introduction
Neurodegeneration with brain iron accumulation (NBIA) is a heterogeneous group of genetic disorders affecting both children and adults with the unifying feature of iron accumulation in specific brain structures, especially globus pallidus. Main defective proteins in different NBIAs have no known direct role in iron homeostasis, but similar pathologic changes in consistent brain areas, mostly basal ganglia, have led to this assumption that iron accumulation is a common pathophysiologic feature among NBIAs.
Different types of NBIA are recognized, labeled according to the molecular finding, replacing the historical term Hallervorden–Spatz disease. The most common subtype is pantothenate kinase-associated neurodegeneration (PKAN) designating the defective enzyme discovered to cause it. Soon after PKAN was described, other malfunctioning enzymes and their responsible genes were discovered and the NBIA group began to expand, today encompassing at least 10 distinct disorders (Table 1). Reference Louis, Mayer and Rowland1–Reference Marras, Lang and van de Warrenburg4 This category is expected to divide further into more subcategories, as still a substantial proportion of “sporadic disorders of uncertain origin” continue to exist as an unclassified heterogeneous subclass of NBIAs. Reference Marras, Lang and van de Warrenburg4
Table 1: The distinct disorders and their responsible proteins and genes comprising the neurodegeneration with brain iron accumulation group

AR = autosomal recessive; Chr = chromosome; HSP = hereditary spastic paraparesis; INAD = infantile neuroaxonal dystrophy; Inher = inheritance; MDS = Movement Disorder Society; NBIA = neurodegeneration with brain iron accumulation; OMIM = Online Mendelian Inheritance in Man; PARK = Parkinson’s disease; SENDA = static encephalopathy of childhood with neurodegeneration in adulthood; SPG = spastic gait gene.
NBIA disorders are rare with an overall estimated prevalence of 1 in 500,000. Their clinical manifestations comprise a wide range of the spectrum (Table 2) mainly movement disorders (especially dystonia and Parkinsonism) but also neuropathy, retinopathy, optic neuropathy, oculomotor abnormalities, mental disability, behavioral, and systemic disorders. Reference Louis, Mayer and Rowland1–Reference Marras, Lang and van de Warrenburg4 Seizure as a clinical manifestation is known to occur in some NBIAs such as phospholipase A2-associated neurodegeneration (PLAN) and beta-propeller protein-associated neurodegeneration (BPAN), but the exact prevalence of epilepsy in each individual disorder is not well elucidated. Reference Hayflick, Kurian and Hogarth2
Table 2: Clinical and brain MRI characteristics of NBIAs

MRI = magnetic resonance imaging.
We reviewed the literature with a systematic method to establish the frequency of seizures reported in NBIA disorders as well as to determine the associated features of patients with seizure.
Method
Search Strategy
All details about search methodology regarding databases, keywords, time and language limits, and inclusion criteria were considered. Reference Ferrari5 The electronic bibliographic databases Pubmed, Scopus, Embase, and Google Scholar were searched for all cases in any type of article from inception to December 16, 2019. The medical subject headings words “seizures” and “epilepsy” and keywords “neurodegeneration with brain iron accumulation,” “Idiopathic NBIA,” “Seitelberger disease,” “infantile neuroaxonal dystrophy” (INAD), “Woodhouse–Sakati syndrome” (WSS), “Kufor–Rakeb syndrome” (KRS), “beta-propeller protein-associated neurodegeneration” (BPAN), “PLA2G6-associated neurodegeneration” (PLAN), “hereditary aceruloplasminemia” (HA), “fatty acid hydroxylase-associated neurodegeneration” (FAHN), “fatty acid 2-hydroxylase” (FA2H), “pantothenate kinase-associated neurodegeneration” (PKAN), “mitochondrial membrane-associated neurodegeneration” (MPAN), and “static encephalopathy of childhood with neurodegeneration in adulthood” (SENDA) were used for the search.
Endnote software X8 was used for the review and inclusion of studies.
Eligibility Criteria
Title and abstract screening and full-text screening were conducted independently by two reviewers. Case reports with an explicit diagnosis of any types of NBIA which were assessed for the incidence of any type of seizure or epilepsy were included. Further cases were identified via cross-referencing between papers. The articles, which were not available in full text or have not been written in English, were excluded. Detailed reasons for the exclusion of papers in each step are provided in the PRISMA diagram (Figure 1).

Figure 1: PRISMA diagram.
Quality Assessment
Two reviewers assessed the quality of the reports using the methodological quality of case reports/series tool. Reference Murad, Sultan, Haffar and Bazerbachi6 This tool proposes an approach to evaluate the quality based on the domains of selection, ascertainment, causality, and reporting. Any disagreements were settled through discussion with a third reviewer.
Data Extraction
Two reviewers independently screened the titles and abstracts of all records. Then, the selection of full texts was accomplished based on the eligibility criteria. The reviewers independently extracted data on study characteristics, type of NBIA and demographic information, clinical characteristics, in particular seizure data including incidence rate, type of seizure, and age of seizure onset. Afterward, a third reviewer cross-checked the extracted data. In order not to miss any cases, we also reviewed cases in the references of all papers, including reviews, but during cross-checking, we ensured to omit the duplicate cases.
Statistical Analysis
All results presented as frequencies and percentages of reported data using Excel® 2016.
Results
A total of 1698 articles was retrieved from the electronic database search. Through the process of duplicate record elimination, title and abstract screening, and eligibility assessments, 71 articles fulfilled the inclusion criteria. After the full-text screening, 51 studies comprising of 305 cases were included (Figure 1, Table 3).
Table 3: Studies’ characteristics, sorted alphabetically by author
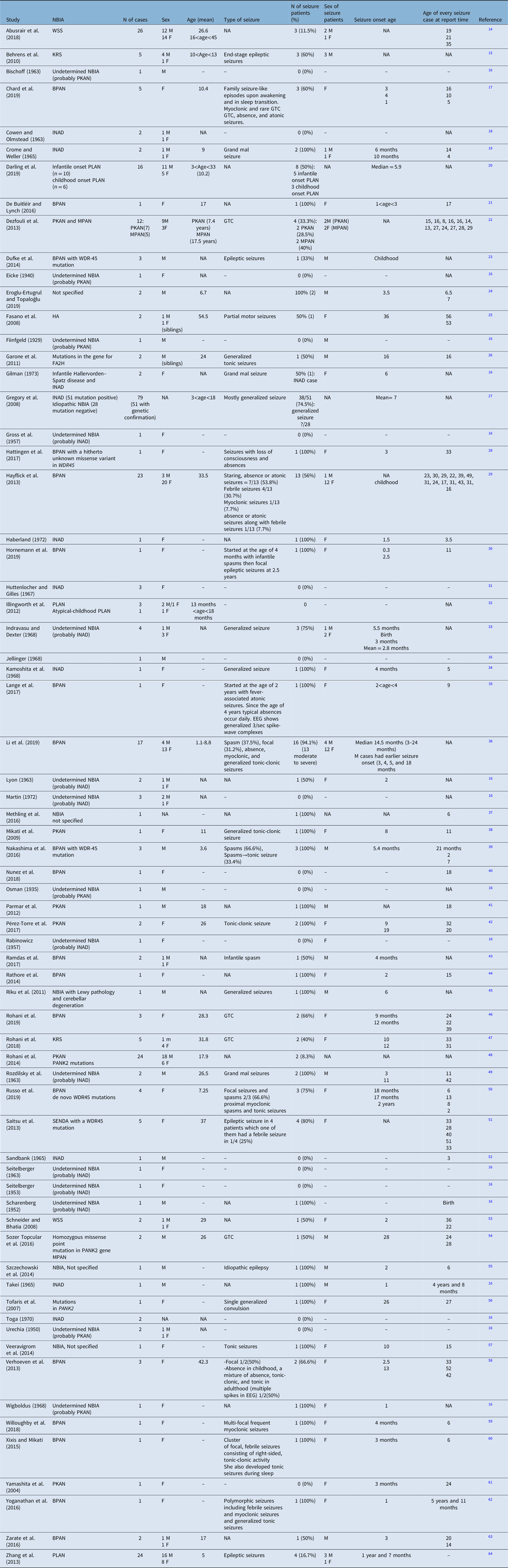
BPAN = beta-propeller protein-associated neurodegeneration; EEG = electroencephalography; FA2H = fatty acid 2-hydroxylase; GTC = generalized tonic-clonic; HA = hereditary aceruloplasminemia; INAD = infantile neuroaxonal dystrophy; KRS = Kufor–Rakeb syndrome; MPAN = mitochondrial membrane-associated neurodegeneration; NBIA = neurodegeneration with brain iron accumulation; PKAN = pantothenate kinase-associated neurodegeneration; PLAN = PLA2G6-associated neurodegeneration; SENDA = static encephalopathy of childhood with neurodegeneration in adulthood; WSS = Woodhouse–Sakati syndrome.
Among these 305 cases in whom the seizure was mentioned as an investigated symptom, 150 (49.2%) had seizures. The incidence of seizure depending on the subtype of NBIA and the demographic characteristics of patients with seizures are presented in Table 4. To summarize, among all NBIA syndromes, seizures were most frequently reported in PLAN (64/126, 50.8%) followed by BPAN (57/79, 72.1%), PKAN (11/47, 23.4%), WSS (4/28, 14.3%), KRS (5/10, 50%), MPAN (2/5, 40%), HA (1/2, 50%), and FAHN (1/2, 50%).
Table 4: The prevalence of epilepsy in each NBIA disorder and the available features related to these patients

BPAN = beta-propeller protein-associated neurodegeneration; FA2H = fatty acid 2-hydroxylase; GTC = generalized tonic-clonic; HA = hereditary aceruloplasminemia; INAD = infantile neuroaxonal dystrophy; KRS = Kufor–Rakeb syndrome; MPAN = mitochondrial membrane-associated neurodegeneration; NBIA = neurodegeneration with brain iron accumulation; PKAN = pantothenate kinase-associated neurodegeneration; PLAN = PLA2G6-associated neurodegeneration; SENDA = static encephalopathy of childhood with neurodegeneration in adulthood; WSS = Woodhouse–Sakati syndrome.
Patients with seizures were more likely to be females and the mean age of seizure onset ranged from 2 to 36 years. The most frequent seizure type in NBIA patients was generalized tonic-clonic seizure, although in BPAN a variety of other seizure types (including infantile spasms, absence, atonic, febrile, myoclonic, focal, proximal myoclonic spasms, and tonic seizures) and in HA focal motor seizure had been reported. However, we should emphasize that most of these papers had been published before the new classification of epilepsy became accessible. So, the type of seizure is based on the previous version of the classification of epilepsy when these papers were released. In most instances, except in some cases of BPAN, seizure was not an early symptom of NBIA. The data regarding the natural history of seizures per se is scarce in the literature. Most of these patients were controlled with one or two antiepileptic drugs.
Discussion
Epilepsy is a frequent neurological disorder, affecting 0.7% of the general population (lifetime prevalence per person is 7/1000 Reference Fiest, Sauro and Wiebe7 ). Causes of seizure in childhood and young patients in order of frequency are genetic (including hereditary neurodegenerative disorders), structural lesions, metabolic disorders, and infections. Reference Sokka, Olsen and Kirjavainen8 In the late-onset sporadic neurodegenerative disorders, such as Alzheimer’s disease and Parkinson’s disease, seizures are reported to occur in about 4.1% and 2.6% of cases, respectively. Reference Vossel, Beagle and Rabinovici9,Reference Feddersen, Rémi, Einhellig, Stoyke, Krauss and Noachtar10 In early-onset neurodegenerative disease, seizures occur more frequently and further complicate the nature of these disorders. One example is NBIA, a group of syndromes clinically characterized by the combination of dystonia, parkinsonism, retinal or optic nerve pathology, and other neurological signs including seizure. Given the lack of systematic data about seizure frequency and characteristics, Reference Hayflick, Kurian and Hogarth2,Reference Vercueil11 we systematically reviewed the literature. Our systematic review revealed that NBIAs as a subcategory of hereditary neurodegenerative disorders are an important cause of epilepsy in children and young adults. Seizure in NBIAs is generally more frequent compared to Wilson’s disease, another metal storage disease, where seizures have been reported in 6.2% of cases. Reference Vercueil11,Reference Dening, Berrios and Walshe12
Among the NBIA disorders, the ones most frequently reported to have epilepsy as a consistent manifestation are PLAN (especially the INAD subtype), BPAN, FAHN, and WSS. Reference Hayflick, Kurian and Hogarth2,Reference Marras, Lang and van de Warrenburg4 By contrast, PKAN – the most common NBIA – is thought to be an infrequent cause of epilepsy. Reference Hayflick, Kurian and Hogarth2
In the present study, in each NBIA disorder, the proportion of patients with seizures was reported to be between 14% (for WSS) and 72% (for BPAN). However, these numbers are biased due to exclusion of papers in which the occurrence of seizure has not been reported and also due to the limited number of case reports for some NBIAs, such as FAHN and HA. These considerations make the estimation of the proportion of patients presenting with seizure very fragile and highlight the importance of complete clinical descriptions, for example, the total number of reported cases of FAHN in papers which alluded to the presence or absence of seizure was two, one of them had seizures, resulting in 50% seizure prevalence in this disorder. Obviously, these percentages cannot be compared to the percentages of more frequent NBIAs such as PLAN with 126 reported cases with seizure annotation in the papers, 64 of them having seizures, resulting in a prevalence of 50% in this disorder. The obvious fact, however, is that PLAN is the most reported NBIA (considering crude numbers) with seizures and this is less biased by the low prevalence, as this disease accounts for nearly 20% of all NBIAs. The other interesting finding of this review is that seizure is not an infrequent finding in PKAN, as 23% of 47 reported PKAN patients happened to be suffering from epilepsy. The other salient result from the current review is that seizure occurrence is more frequent in BPAN (72%) than in any other NBIA and this seems to be a solid result. In spite of the lower prevalence of BPAN compared to PLAN (2% vs. 20% of all NBIAs), the crude number of BPAN patients with seizures stand just next to PLAN (57 and 64 cases, respectively). We estimate based on the present study that BPAN is the most probable NBIA with seizure included in the clinical picture (nearly 72%) followed by PLAN (50%) and PKAN (23%). The other NBIAs, which may be commonly accompanied by seizures, would be WSS, KRS, MPAN, FAHN, and HA, but the exact proportion of patients with seizures and the order of frequency among these disorders cannot be judged with available data.
As already outlined, the findings of our systematic need to be interpreted with caution as no study were specifically addressed to screen the NBIA population for the occurrence of seizures. Our assumptions are indeed based on individual case reports and case series. In addition, the data regarding the natural history of seizures and treatment response were scarce in the literature, thus making it hard to draw conclusions also in terms of the relationship with the natural history of these progressive disorders. Likewise, the data regarding seizure onset age, sex, seizure type, and phenomenology was insufficient, and the results might be distorted by excessive missing data. On the other hand, one cannot be sure that all the reported seizures in the previous reports have been epileptic in nature, for example, the possibility of including provoked seizures should be considered as a bias which cannot be sorted out retrospectively. In addition, the papers were from a diverse time period and almost all of them were published before the new classification of epilepsy. Therefore, the new classification could not be applied to the cases. Reference Fisher, Cross and French13 For instance, based on these data, we were not able to differentiate between generalized tonic-clonic seizures and bilateral tonic-clonic ones which is a new concept. Another problem regarding the classification was that the seizure type, which was not the goal of most reports, was not mentioned or clearly understandable from the reports (see Table 3). So, the readers are advised to consider this issue as a major limitation. Another limitation to our review that readers should be aware of is that this study solely reviewed the English literature on NBIAs which can make the conclusion more biased.
Nevertheless, our study is the first study in this regard, and future studies with more precise information will be needed to clarify this issue.
Disclosures
ME, PS, NM, and MR have no disclosures. SAS was supported by the LMU Clinician Scientist Programme and the Stiftung Verum, receives royalties from Thieme Publisher, receives consulting fees from Medavante Prophase, and receives honoraria from Movement Disorders Society. AF receives consultancy fees from Apple, Abbvie, Abbott, Medtronic, Boston Scientific, Sunovion, Chiesi farmaceutici, UCB, Ipsen, and Rune Labs, receives honoraria from the American Academy of Neurology, Abbott, Abbvie, Medtronic, Boston Scientific, Sunovion, Chiesi farmaceutici, UCB, Ipsen, Paladin Lab, and Movement Disorders Society, receives grant support from the University of Toronto, Abbvie, Medtronic, Boston Scientific, MSA Coalition, and McLaughlin Centre, and receives royalties from springer and sits on advisory boards for Abbvie, Boston Scientific, and Ipsen.
Authors Contributions
ME: writing of the first draft; PS: design of the study and writing of the first draft; SAS: review and critique; AF: conception, review, and critique; NM: review and critique; MR: conception, design of the study, review, and critique.





