


Humans are continuously exposed to a wide array of environmental toxicants including organic chemicals. 1,1,2-Trichloroethylene (TCE) is a widespread organic solvent used in degreasing metals and in ink and varnishes. It is also an intermediate product in the synthesis of fluorochemicals. TCE is not found naturally, but has been found in measurable amounts in the food chain and in drinking water. During production, storage and use, TCE evaporates into the atmosphere and has been identified as a common air contaminant. In addition, leakage from chemical waste sites has led to its contamination of groundwater(Reference Lock and Reed1). The maximum permissible level of TCE in drinking water according to the United States Environmental Protection Agency is 5 parts per billion (5 μg/l). Recently, the United States Environmental Protection Agency(2) and the International Agency for Research on Cancer have designated TCE as ‘carcinogen to humans’ by all routes of exposure. According to published reports, the kidneys and liver are the most affected target organs after TCE exposure in humans and animals(Reference Bull, Orner and Cheng3, Reference Lash, Fisher and Lipscomb4). Further, acute and chronic exposure of TCE results in toxicity (induced pathology) in a variety of organs in humans and experimental animals(Reference Lock and Reed1, Reference Ruder5).
The bulk of TCE biotransformation occurs in the liver, though metabolic activation of relatively small quantities of TCE reaching extra-hepatic tissues, such as kidneys, testes and lungs, can have toxicologically significant impacts in situ (Reference Lash, Fisher and Lipscomb4, Reference Forkert, Lash and Nadeau6, Reference Forkert, Millen and Lash7). The toxicity of TCE is dependent on its bioactivation, which occurs either by glutathione (GSH) conjugation in the liver to form dichlorovinyl GSH or by cytochrome (P450)-dependent oxidation to form dichlorovinyl cysteine. Dichlorovinyl GSH is either further metabolised in the kidney to the cysteine conjugate, which is metabolically activated to a thioacetylating agent by β-lyase, or detoxified by N-acetyltransferase and released in the urine(Reference Lash, Fisher and Lipscomb4). Dichlorovinyl cysteine can also undergo sulphoxidation to form a reactive intermediate(Reference Sausen and Elfarra8). Another metabolite of TCE is formed as a result of flavin-containing mono-oxygenase-mediated sulphoxidation of dichlorovinyl cysteine. Dichlorovinyl cysteine sulphoxide has been reported to be nephrotoxic in rats and also cytotoxic to isolated rat kidney cells(Reference Lash9). TCE has been shown to have a toxic effect on carbohydrate metabolism and brush-border enzymes in the rat kidney(Reference Khan, Priyamvada and Khan10), as well as damaging effects on skeletal muscles(Reference Vattemi, Tonin and Filosto11). TCE can induce oxidative stress-mediated apoptosis in humans as well as animal models. For example, Chen et al. (Reference Chen, Wang and Chen12) showed TCE-mediated induction of apoptosis in human lung cancer cells. Similarly, Lash et al. (Reference Lash, Putt and Hueni13) also reported the induction of apoptosis in a culture of human renal cortical cells.
Recently, researchers all over the world have focused their interest on the isolation, elucidation and effectiveness of naturally occurring anti-inflammatory and anticancer agents that are usually active ingredients of the daily diet(Reference Nobili, Lippib and Witortc14). The positive health effects from the consumption of a diet rich in fruits and vegetables are mainly due to the presence of antioxidants such as carotenoids, polyphenols and anthocyanins. These molecules target the expression of multiple genes and proteins involved in tumour promotion cell signalling(Reference Nobili, Lippib and Witortc14). Minor dietary non-nutrients such as flavonoids, derived from flavones with various degrees of hydroxylation and glycosidic substitutions, are showing promising results as potential chemopreventive agents(Reference Kaefer and Milner15).
Flavonoids are natural polyphenols ubiquitously present in many plants(Reference Wang and Morris16). Epidemiological studies have advocated that dietary supplements rich in flavonoids prevent various diseases, namely cancers, diabetes, CVD and neurodegenerative diseases(Reference Clere, Faure and Martinez17–Reference Mandel, Amit and Weinreb20). Diosmin (diosmetin 7-O-rutinoside), a naturally occurring flavone glycoside readily obtained by the dehydrogenation of hesperidin, is found abundantly in the pericarp of various citrus fruits(Reference Campanero, Escolar and Garcia-Quetglas21). Diosmin is a biologically very active polyphenol and has been shown to possess antioxidant, anti-inflammatory, antidiabetic, anti-atherosclerotic and anti-apoptotic activities(Reference Galati, Monforte and Kirjavainen22–Reference Manuel, Keenoy and Vertommen27). Tanaka et al. (Reference Tanaka, Makita and Ohnishi28–Reference Tanaka, Makita and Kawabata30) reported the protective efficacy of diosmin against N-methyl-N-amylnitrosamine-induced oesophageal tumorigenesis, azoxymethane-induced rat colon carcinogenesis and 4-nitroquinoline 1-oxide-induced oral carcinogenesis.
In light of the above inferences from other evidence, we designed the present study to investigate the protective effects of diosmin against TCE-induced renal toxicity and apoptotic response in the normal renal tissue of Wistar rats.
Materials and methods
Antibodies and reagents
TCE was purchased from Merck, while all other chemicals and reagents were procured from Sigma-Aldrich Company, unless otherwise stated. Antibodies p53 and Bax were procured from Santa Cruz Biotechnology, Inc. and Abcam, respectively.
Animals
Young (8–10 weeks old), male Wistar rats were housed in plastic (polypropylene) cages in an animal house facility at Hamdard University. The experiments were conducted according to protocols approved by the Committee for the Purpose of Control and Supervision of Experimental Animals (CPCSEA), New Delhi, India (project no. 547/CPCSEA, 28 May 2009). The well-ventilated animal rooms (room temperature set at 25°C) were maintained on 12 h light—12 h dark cycles. They were acclimatised for 1 week before the study and had free access to a standard laboratory feed (Hindustan Lever Limited) and water ad libitum.
Experimental design
For the study of biochemical parameters and immunohistochemistry, thirty male Wistar rats were divided into five groups of six animals each. Group I served as a vehicle-treated control and was given an oral treatment of maize oil for only 20 d. Group II served as a treatment control and was given an oral administration of TCE for 20 d (1000 mg/kg body weight), freshly dissolved in maize oil. Diosmin was orally administered at two doses, 20 (D1) and 40 (D2) mg/kg body weight, to groups III and IV, respectively, for twenty consecutive days. Groups III and IV also received an oral treatment of TCE (1000 mg/kg body weight) consecutively for 20 d. Group V only received D2 of diosmin for twenty consecutive days. All the animals were killed after 12 h after the last dose of TCE (Fig. 1). The kidneys were removed and processed for enzyme estimation and other parameters. The above-mentioned doses of diosmin were selected based on preliminary studies, while the dose of TCE was selected in accordance with the report of Khan et al. (Reference Khan, Priyamvada and Khan10).

Fig. 1 Schematic representation of the experimental design. BW, body weight; TCE, trichloroethylene. A colour version of this figure can be found online at http://www.journals.cambridge.org/bjn
DNA isolation
DNA extraction was done by the standard chloroform–isoamyl method. DNA was extracted from approximately 400 mg of kidney tissue by homogenising the tissue in 5 ml TNE buffer (50 mm-Trisma, 100 mm-EDTA, 0·5 % SDS, pH 8·0) in a 2 ml ground glass homogeniser. Each sample was homogenised with ten standardised strokes of the pestle to minimise any potential effect on DNA integrity introduced by the homogenisation procedure. An equal volume of buffered phenol–chloroform–isoamyl alcohol (25:24:1, by vol., pH 8·0) was then added to the sample. The sample was gently mixed and allowed to settle for 5 min and then centrifuged for 5 min at 13 000 rpm at 4°C. The aqueous layer was transferred to a new microcentrifuge tube and phenol–chloroform–isoamyl alcohol extraction was repeated. The aqueous layer was then digested by 5 ml RNase (10 mg/ml) for 30 min at 37°C, and the digest was extracted once by phenol–chloroform–isoamyl alcohol and once by 500 ml chloroform. DNA was precipitated from the resulting aqueous layer by adding two volumes of absolute ethanol and one-tenth volume of 3 m-sodium acetate (pH 5·2). The sample was then centrifuged (13 000 rpm, 15 min), and the resulting pellet rinsed with 500 ml of 70 % ethanol and air-dried. The amount of DNA was quantified spectrophotometrically at 260 and 280 nm. Then, 2 mg/ml of DNA sample was dissolved in 1 ml of TE buffer (10 mm-Trisma, 1 mm-EDTA) and subsequently used for the next experiment.
Alkaline unwinding assay (DNA strand breaks)
In the alkaline unwinding assay, the rate of transition of double-stranded DNA (dsDNA) to single-stranded DNA (ssDNA) under a predefined alkaline denaturing condition was proportional to the number of breaks in the phosphodiester backbone, and thus was used as a measure of DNA integrity. Bisbenzamide was used as a DNA-binding dye from the fluorescence of which various types of DNA were quantified. For the fluorescence determination of dsDNA, ssDNA and partially unwound DNA, three equal portions of diluted DNA sample were prepared. The amount of dsDNA was obtained from the fluorescence of a sample without any treatment; while ssDNA was determined from the sample that had been boiled for 30 min. Fluorescence of the DNA sample that had been subjected to alkaline treatment (pH 12·2) on ice for 30 min provided an estimate of the amount of unwound DNA. The fluorescence of initial or dsDNA was determined by placing 100 μmol DNA sample, 100 μl NaCl (25 mm) and 2 μl SDS (0·5 %) in a pre-chilled test tube, followed by the addition of 3 ml of 0·2 m-potassium phosphate (pH 9) and 3 μl bisbenzamide (1 mg/ml). The contents were mixed and allowed to react in the dark for 15 min to allow the fluorescence to stabilise. The fluorescence of the sample was then measured using a spectrofluorometer (excitation 360 nm; emission 450 nm). The fluorescence of ssDNA was determined as above but by using the DNA sample that had already been boiled for 30 min to completely unwind the DNA. Thereafter, 50 μl NaOH (0·05 m) was rapidly mixed with 100 μl of the DNA sample in a pre-chilled test tube. The mixture was incubated on ice in the dark for 30 min followed by the rapid addition and mixing of 50 μl HCl (0·05 m). This was followed immediately by an addition of 2 μl SDS (0·5 %) and the mixture was forcefully passed through a 21 G needle five times. The fluorescence of alkaline unwound DNA sample was measured as described above. Measurement of the alkaline unwound sample was performed in triplicate and the average was reported. The ratio between dsDNA and total DNA (F value) was determined as follows:
 $$\begin{eqnarray} F \,value = (auDNA - ssDNA)/(dsDNA - ssDNA), \end{eqnarray}$$
$$\begin{eqnarray} F \,value = (auDNA - ssDNA)/(dsDNA - ssDNA), \end{eqnarray}$$
where auDNA, ssDNA and dsDNA are the degrees of fluorescence from the partially unwound, single-stranded and double-stranded determinations, respectively. The F value was inversely proportional to the number of strand breaks present, and thus could be used as an indicator of DNA integrity.
Post-mitochondrial supernatant preparation and biochemical estimations
Post-mitochondrial supernatant (PMS) was prepared by the method of Tahir & Sultana(Reference Tahir and Sultana31), with some modifications. In brief, the kidneys were removed quickly, cleaned of extraneous material and immediately perfused with ice-cold saline (0·85 % NaCl). The kidneys were homogenised in chilled phosphate buffer (0·1 m, pH 7·4) containing KCl (1·17 %) using a Potter–Elvehjen homogeniser. The homogenate was filtered through muslin cloth and centrifuged at 800 g for 5 min at 4°C by a REMI cooling centrifuge to separate the nuclear debris. The aliquot obtained was centrifuged at 12 000 rpm for 20 min at 4°C to obtain the PMS, which was used as a source of enzymes. All biochemical estimations were completed within 24 h of animal killing.
Assay for catalase activity
Catalase activity was assessed by the method of Claiborne(Reference Claiborne and Greenwald32). In short, the reaction mixture consisted of 0·05 ml PMS, 1·0 ml of H2O2 (0·019 m), 1·95 ml phosphate buffer (0·1 m, pH 7·4), in a total volume of 3 ml. Changes in absorbance were recorded at 240 nm, and the change in absorbance was calculated as nmol H2O2 consumed/min per mg protein.
Estimation of lipid peroxidation
The assay of lipid peroxidation (LPO) was done according to the method of Wright et al. (Reference Wright, Colby and Miles33). The reaction mixture consisted of 0·58 ml phosphate buffer (0·1 m, pH 7·4), 0·2 ml microsome, 0·2 ml ascorbic acid (100 mm) and 0·02 ml ferric chloride (100 mm), in a total volume of 1 ml. This reaction mixture was then incubated at 37°C in a shaking water bath for 1 h. The reaction was stopped by the addition of 1 ml trichloroacetic acid (10 %). Following the addition of 1·0 ml thiobarbituric acid (TBA) (0·67 %), all the tubes were placed in a boiling water bath for a period of 20 min. The tubes were shifted to an ice bath and then centrifuged at 2500 g for 10 min. The amount of malondialdehyde (MDA) formed in each of the samples was assessed by measuring the optical density of the supernatant at 535 nm. The results were expressed as nmol TBA formed/h per g tissue at 37°C by using a molar extinction coefficient of 1·56 × 105/ m per cm.
Estimation of glutathione
GSH was assessed by the method of Jollow et al. (Reference Jollow, Mitchell and Zampaglione34). A quantity of 1·0 ml of 10 % PMS mixed with 1·0 ml of (4 %) sulphosalicylic acid was taken, incubated at 4°C for a minimum time period of 1 h and then centrifuged at 4°C at 1200 g for 15 min. The reaction mixture of 3·0 ml was composed of 0·4 ml of supernatant, 2·2 ml phosphate buffer (0·1 m, pH 7·4) and 0·4 ml dithio-bis-2-nitrobenzoic acid (4 mg/ml). The yellow colour developed was read immediately at 412 nm on the spectrophotometer (Lambda EZ201; Perkin Elmer). GSH concentration was calculated as nmol GSH conjugates/g tissue.
Measurement of superoxide dismutase activity
Superoxide dismutase (SOD) activity was measured by the method of Marklund & Marklund(Reference Marklund and Marklund35). The reaction mixture consisted of 2·875 ml Tris–HCl buffer (50 mm, pH 8·5), pyrogallol (24 mm in 10 mm-HCl) and 100 μl PMS, in a total volume of 3 ml. Enzyme activity was measured at 420 nm and was expressed as units/mg protein. One unit of enzyme is defined as the enzyme activity that inhibits the auto-oxidation of pyrogallol by 50 %.
Assay for glutathione peroxidase activity
The activity of GSH peroxidase (GPx) was calculated by the method of Mohandas et al. (Reference Mohandas, Marshall and Duggin36). The total volume of 2 ml was composed of 0·1 ml EDTA (1 mm), 0·1 ml sodium azide (1 mm), 1·44 ml phosphate buffer (0·1 m, pH 7·4), 0·05 ml glutathione reductase (1 IU/ml is equivalent to 1 mol GSSG reduced/min per ml), 0·05 ml GSH (1 mm), 0·1 ml NADPH (0·2 mm), 0·01 ml H2O2 (0·25 mm) and 0·1 ml of 10 % PMS. The depletion of NADPH at 340 nm was recorded at 25°C. Enzyme activity was calculated as nmol NADPH oxidised/min per mg protein with a molar extinction coefficient of 6·22 × 103/m per cm.
Glutathione reductase activity
GR activity was determined by the method of Carlberg & Mannervik(Reference Carlberg and Mannervik37). The reaction mixture consisted of 1·65 ml phosphate buffer (0·1 m, pH 7·6), 0·1 ml EDTA (0·5 mm), 0·05 ml GSH (1 mm), 0·1 ml NADPH (0·1 mm) and 0·1 ml of 10 % PMS, in a total volume of 2 ml. Enzyme activity was quantified at 25°C by measuring the disappearance of NADPH at 340 nm and was calculated as nmol NADPH oxidized/min per mg protein using a molar extinction coefficient of 6·22 × 103/m per cm.
Glutathione S-transferase activity
The reaction mixture consisted of 2·5 ml phosphate buffer (0·1 m, pH 6·5), 0·2 ml GSH (1 mm), 0·2 ml 1-chloro-2,4-dinitrobenzene (1 mm) and 0·1 ml of the cytosolic fraction (10 %), in a total volume of 3·0 ml. Changes in absorbance were recorded at 340 nm, and enzymatic activity was calculated as nmol 1-chloro-2,4-dinitrobenzene conjugate formed/min per mg protein using a molar extinction coefficient of 9·6 × 103/m per cm(Reference Habig, Pabst and Jakoby38).
Blood urea nitrogen
Estimation of blood urea N (BUN) was done by the method of Kanter(Reference Kanter39). A protein-free filtrate was prepared by adding serum and an equal amount of 10 % trichloroacetic acid, and then the mixture was centrifuged at 2000 rpm and the supernatant was obtained. To 0·5 ml of the protein-free filtrate were added 3·5 ml of distilled water, 0·8 ml diacetylmonoxime (2 %) and 3·2 ml H2SO4–phosphoric acid reagent (reagent was prepared by mixing 150 ml of 85 % phosphoric acid with 140 ml of water and 50 ml of concentrated H2SO4). The reaction mixture was placed in a boiling water bath for 30 min and then cooled. Absorbance was read at 480 nm.
Serum creatinine level
Creatinine was estimated by the method of Hare(Reference Hare40). A protein-free filtrate was prepared. To 1·0 ml of serum were added 1·0 ml sodium tungstate (5 %), 1·0 ml H2SO4 (0·6 m) and 1·0 ml of distilled water. After mixing thoroughly, the mixture was centrifuged at 800 g for 5 min. The supernatant was added to a mixture containing 1·0 ml picric acid (1·05 %) and 1·0 ml NaOH (0·75 m). Absorbance at 520 nm was read exactly after 20 min.
Assay for lactate dehydrogenase activity
Lactate dehydrogenase (LDH) activity was estimated in serum by the method of Kornberg(Reference Kornberg41). The assay mixture consisted of 0·2 ml of serum, 0·1 ml of 0·02 m-NADH, 0·1 ml of 0·01 m-sodium pyruvate, 1·1 ml of 0·1 m (pH 7·4) phosphate buffer and distilled water, in a total volume of 3 ml. Enzyme activity was recorded at 340 nm, and activity was calculated as nmol NADH oxidised/min per mg protein.
Estimation of protein
Protein concentration in all the samples was determined by the method of Lowry et al. (Reference Lowry, Rosebrough and Farr42), using bovine serum albumin as the standard.
Estimation of caspase
Caspase-3, 7 and 9 levels were measured with a kit (CasPASE Assay Kit; G-Bioscience) according to the manufacturer's directions. Results were expressed as μmol/mg protein.
Estimation of kidney injury molecule-1
Kidney injury molecule-1 (KIM-1) levels were measured by a commercially available ELISA-based kit (RAT KIM-1 ELISA Kit; Adipo Bioscience, Inc.) following the manufacturer's instructions.
Immunohistochemistry
The processed renal tissues were obtained and preserved in 10 % paraformaldehyde overnight followed by dehydration in 30, 20 and 10 % sucrose solution successively up to 3 d, and were then fixed in formaldehyde fixative until immunochemical staining. Then, 5–15 μm-thick sections of paraffin-embedded tissues were cut using a grading-type Leica microtome and boiled in 0·1 m-citrate buffer (pH 6·0) for 5 min for the antigen retrieval process, and then incubated in 0·3 % H2O2 in methanol followed by incubation in blocking buffer containing 0·1 m-PBS, 0·04 % Triton X-100 and 10 % normal goat serum. Tissue sections were stained with antibodies against Bax (1:100 dilution) and anti-p53 antibody (1:200) overnight at 4°C. After rinsing in the buffer, the sections were processed using a three-layer peroxidase staining kit (Thermo Scientific System). The peroxide complex was visualised with 3,3-diaminobenzidine. Lastly, the slides were counterstained with haematoxylin for 5 s. The slides were then cleaned in sterile HPLC-grade water supplied by BDH, gradually dehydrated with ethanol, coverslipped in mounting medium and photographed under an Olympus microscope (BX51).
Statistical analysis
The data from individual groups are presented as means with their standard errors. Differences between groups were analysed by using ANOVA followed by the Tukey–Kramer multiple comparison test. The minimum criterion for statistical significance was set at P< 0·05 for all comparisons.
Results
Trichloroethylene exposure leads to the depletion of renal antioxidants but amelioration with diosmin
TCE administration caused a significant decrease in the activities of GSH-metabolising enzymes, namely GST (P< 0·001, 53·8 %), GPx (P< 0·001, 52·9 %) and GR (P< 0·001, 50·4 %) when compared with the control group (Table 1). Diosmin significantly and dose-dependently recovered the activities of GST (D1, P< 0·01, 29·3 %; D2, P< 0·001, 70·9 %), GPx (D1, P< 0·01, 43·3 %; D2, P< 0·001, 58·2 %) and GR (D1, P< 0·01, 35·4; D2, P< 0·001, 78·7 %) antioxidant enzymes. No significant change was observed with diosmin-only-treated animals when compared with the control. GSH data also showed a significant decrease (P< 0·001, 46·6 %) in animals treated with TCE and the treatment with diosmin significantly recovered GSH levels dose-dependently (D1, P< 0·05, 25 %; D2, P< 0·001, 62·5 %). Furthermore, the exhaustive depletion of GSH-dependent enzymes led to the disruption of other oxidative stress signalling cascade enzymes such as the decrease in SOD (P< 0·001, 51·3 %) and catalase (P< 0·001, 65 %) activity (Table 3). The diosmin treatment was effective in increasing the levels of SOD (D1, P< 0·05, 30·2 %; D2, P< 0·001, 63·3 %) and catalase (D1, P< 0·05, 47·3 %; D2, P< 0·001, 146·3 %) to normal at both the doses.
Table 1 Effect of the pretreatment of diosmin (DM) on the antioxidant enzymes glutathione (GSH), glutathione S-transferase (GST), glutathione reductase (GR) and glutathione peroxidase (GPx) on trichloroethylene (TCE)-induced renal redox imbalance (Mean values with their standard errors and percentage changes)

CDNB, 1-chloro-2,4-dinitrobenzene; DM 20, 20 mg DM/kg body weight; DM 40, 40 mg DM/kg body weight.
*** Mean value was significantly different from group I (P< 0·001).
Mean values were significantly different from group II: † P< 0·05, †† P< 0·01, ††† P< 0001.
Effect of diosmin on renal membrane damage (lipid peroxidation)
MDA formation in renal PMS was measured to demonstrate oxidative damage in the TCE-induced renal injury of Wistar rats. A significant (P< 0·001, 100·1 %) rise in the level of MDA formation was found in the TCE-treated group in renal tissue when compared with the control. We observed that the treatment with diosmin at both the doses led to the significant restoration (D1, P< 0·01, 35·2 %; D2, P< 0·001, 43·6 %) of membrane integrity in renal tissue when compared with the TCE-treated group (Table 3).
Diosmin treatment inhibits renal damage
The effects of diosmin administration on TCE-mediated elevated levels of kidney toxicity markers (BUN, LDH and creatinine) are shown in Table 2. The animals treated with TCE showed a significant increase in BUN (P< 0·001, 161·5 %), creatinine (P< 0·001, 177 %) and LDH (P< 0·001, 142·3 %) levels when compared with the control. A marked inhibition was observed at both doses in BUN (D1, P< 0·05, 38·0 %; D2, P< 0·001, 44·1 %), creatinine (D1, P< 0·05, 25·6 %; D2, P< 0·001, 46·2 %) and LDH (D1, P< 0·05, 27·7 %; D2, P< 0·01, 46·8 %). No significant difference was found in the D2-only group compared with the control (Table 2).
Table 2 Effect of the pretreatment of diosmin (DM) on the serum markers blood urea nitrogen (BUN), creatinine, lactate dehydrogenase (LDH) and kidney injury molecule-1 (KIM-1) on trichloroethylene (TCE)-induced enhancement (Mean values with their standard errors and percentage changes)

DM 20, 20 mg DM/kg body weight; DM 40, 40 mg DM/kg body weight.
*** Mean value was significantly different from group I (P< 0·001).
Mean values were significantly different from group II: † P< 0·05, †† P< 0·01, ††† P< 0001.
Diosmin treatment inhibits kidney injury molecule-1 level
TCE administration increased the level of KIM-1 in group II compared with group I (P< 0·001, 387·7 %). The diosmin treatment at both the doses decreased the TCE-induced abnormal increase in the level of serum KIM-1 (D1, P< 0·05, 20·7 %; D2, P< 0·01, 50·6 %). There was no significant difference between the control (group I) and diosmin-only-treated groups (group V).
Diosmin decreases the number of DNA strand breaks in the alkaline unwinding assay (marker for DNA damage)
In the DNA alkaline unwinding assay (Table 3), a significant decrease in the F value (P< 0·001, 60·7 %) in the TCE-treated group (group II) was observed compared with the control group (group I), whereas the F value increased at a lower dose (P< 0·05, 39·9 %) and at a higher dose of DM (P< 0·01, 130·3 %).
Table 3 Effect of the pretreatment of diosmin (DM) on malondialdehyde (MDA), superoxide dismutase (SOD), catalase and DNA strand breaks on trichloroethylene (TCE)-induced renal toxicity (Mean values with their standard errors and percentage changes)

DM 20, 20 mg DM/kg body weight; DM 40, 40 mg DM/kg body weight.
*** Mean value was significantly different from group I (P< 0·001).
Mean values were significantly different from group II: † P< 0·05, †† P< 0·01, ††† P< 0001.
Diosmin modulates apoptotic cell death mechanisms
Caspase-3 and -7 are effector caspases and caspase 9 is an initiator caspase. The levels of all these caspases were measured and a steep increase in the activity of all the caspases (P< 0·001) was observed in the TCE-treated group (group II) compared with the control (group III). The diosmin treatment dose-dependently and significantly increased the level of all the caspases studied to normal (Fig. 4).
Genomic instability is basically linked to the change in the control of apoptosis and apoptosis-associated genes, including Bcl-2, Bax, p53 and poly-ADP-ribose polymerase (PARP). The expression of Bax and p53 in renal tissue was examined by immunohistochemistry. We could not detect any Bax and p53 positive region in the kidney of control animals (group I); positive staining of these proteins was located in the cytoplasm of renal cells from the TCE-treated animals (group II). A higher dose of diosmin (group IV) decreased p53 (Fig. 2) and Bax (Fig. 3) immunopositivity when compared with the TCE-treated group (group II). There was no difference in immunoreactivity in the case of the diosmin-only-treated group when compared with the control (data not shown).
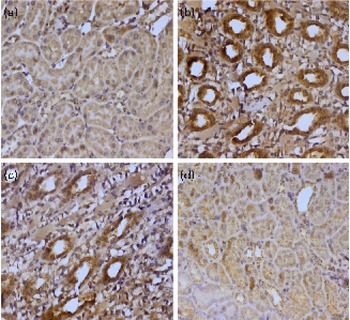
Fig. 2 Effect of diosmin (DM) pretreatment on trichloroethylene (TCE)-induced p53 expression. Photomicrographs of the kidney sections depicting (a) the vehicle-treated control group (group I), (b) the TCE-treated group (1000 mg/kg body weight, group II), (c) dose 1 of DM (20 mg/kg body weight)+TCE (group III) and (d) dose 2 of DM (40 mg/kg body weight)+TCE (group IV). For immunohistochemical analyses, the dark brown colour indicated specific immunostaining of p53 and the light colour indicated nuclear haematoxylin staining. The kidney section of the TCE-treated group (group II) had more p53-immunopositive staining as indicated by the brown colour when compared with the control group (group I), while the pretreatment of DM in groups III and IV reduced p53 immunostaining when compared with group II (40 × magnification). A colour version of this figure can be found online at http://www.journals.cambridge.org/bjn

Fig. 3 Effect of diosmin (DM) co-treatment on trichloroethylene (TCE)-induced Bax expression. Photomicrographs of the kidney sections depicting (a) the vehicle-treated control group (group I), (b) the TCE-treated group (1000 mg/kg body weight, group II), (c) dose 1 of DM (20 mg/kg body weight)+TCE (group III) and (d) dose 2 of DM (40 mg/kg body weight)+TCE (group IV). For immunohistochemical analyses, the intense dark brown colour indicated specific immunostaining of Bax and the light colour indicated nuclear haematoxylin staining. The kidney section of the TCE-treated group (group II) had more Bax-immunopositive staining as indicated by the brown colour when compared with the control group (group I), while the pretreatment of DM in groups III and IV reduced Bax immunostaining when compared with group II (40 × magnification). A colour version of this figure can be found online at http://www.journals.cambridge.org/bjn
Effects of diosmin and trichloroethylene on the histology of the kidney
The haematoxylin and eosin-stained sections exhibited normal histo-architecture in the control group while the TCE-treated group showed distorted tubular architecture, vacuolar formation, degeneration of tubular architecture, tubular congestion, swelling and blebbing. Diosmin significantly attenuated the TCE-induced histopathological changes at both the doses (20 and 40 mg/kg body weight). There was no significant difference in histological changes in the group V-treated group when compared with group I (Fig. 5).

Fig. 4 Effect of the co-treatment of diosmin (DM) on the trichloroethylene (TCE)-induced modulation of apoptotic proteins caspase-3 (■), caspase-7 (![]() ) and caspase-9 (
) and caspase-9 (![]() ). Values are means, with their standard errors represented by vertical bars. TCE administration resulted in a significant increase in the level of caspase-3, caspase-7 and caspase-9 (***P< 0·001). Co-treatment with DM at both doses significantly modulated the alterations in the level of caspase-3, -7 and -9 induced by TCE in the rat kidney. The DM treatment significantly restored TCE induced over activation of all the caspases studied: † P< 0·05, †† P< 0·01, ††† P< 0·001. DM1, 20 mg DM/kg body weight; DM2, 40 mg DM/kg body weight.
). Values are means, with their standard errors represented by vertical bars. TCE administration resulted in a significant increase in the level of caspase-3, caspase-7 and caspase-9 (***P< 0·001). Co-treatment with DM at both doses significantly modulated the alterations in the level of caspase-3, -7 and -9 induced by TCE in the rat kidney. The DM treatment significantly restored TCE induced over activation of all the caspases studied: † P< 0·05, †† P< 0·01, ††† P< 0·001. DM1, 20 mg DM/kg body weight; DM2, 40 mg DM/kg body weight.

Fig. 5 Effects of diosmin (DM) and trichloroethylene (TCE) on the histo-architecture of the kidney. Photomicrographs of the kidney sections depicting (a) the vehicle-treated control group (group I), (b) the TCE-treated group (1000 mg/kg body weight, group II), (c) dose 1 of DM (20 mg/kg body weight)+TCE (group III) and (d) dose 2 of DM (40 mg/kg body weight)+TCE (group IV). (a) Normal kidney histology from the control rats. (b) TCE administration caused the formation of necrotic, vacuolated swell cells (represented by arrows), glomeruli exhibiting swelling with irregular Bowman's capsular space. (c) The administration of a lower dose of DM (20 mg/kg body weight) partially prevented the cytotoxic damage induced by TCE. (d) A higher dose of DM (40 mg/kg body weight) showed almost complete protection to the rat kidney section from the damage induced by TCE as evident from the normal histology of the inner cortical region. A colour version of this figure can be found online at http://www.journals.cambridge.org/bjn
Discussion
In the present study, we have observed the protective effects of diosmin against TCE-induced renal toxicity in Wistar rats. TCE-induced renal toxicity is well documented(Reference Lock and Reed1, 2). However, the exact mechanism underlying TCE-induced renal toxicity is still unclear, but it may be due to reactive oxygen species generated by TCE which leads to the condition of oxidative stress. Therefore, natural compounds with antioxidant properties are gaining much attention. The present study was carried out to elucidate the effect of diosmin on TCE-induced renal toxicity in rats, and to assess its role in the modulation of the apoptotic pathway.
TCE is a common volatile solvent widely used in the metal degreasing (and in the ink and varnishes) industry. Humans are exposed either through the inhalatory route in the form of TCE vapours or orally as a water contaminant(2). TCE exposure has been associated with a range of adverse health effects such as renal toxicity, hepatotoxicity, neurotoxicity, immunotoxicity, developmental toxicity, endocrine toxicity and numerous forms of cancers(Reference Lock and Reed1, 2, Reference Lash, Fisher and Lipscomb4, Reference Chiu, Caldwell and Keshava43).
Flavonoids, secondary plant metabolites, are non-nutritive but biologically active polyphenolic compounds(Reference Crozier, Burns and Aziz44). Flavonoids are present in a variety of vegetables, fruits and seeds, many of which have been found to exhibit a wide range of biological activities including antioxidant, anti-inflammatory, anti-allergic and anti-tumour effects(Reference Middleton, andaswami and Theoharides45–Reference Beecher47). Diosmin, a naturally occurring flavone, exhibits anti-inflammatory, antioxidant and anti-mutagenic properties(Reference Del Bano, Lorente and Castillo48–Reference Nogata, Sakamoto and Shiratsuchi50). Several investigators have also reported the anti-proliferative effect of diosmin in various animal models and human cancer cell lines(Reference Yang, Tanaka and Hirose51, Reference Kuntz, Wenzel and Daniel52).
Oxidative stress is implicated in nephrotoxicity(Reference Cojocel, Beuter and Müller53). TCE-induced toxicity is mainly dependent on bioactivation, which occurs by two pathways: GSH conjugation and cytochrome P450-dependent oxidation(Reference Lash, Parker and Scott54, Reference Lash, Hueni and Putt55). Major biochemical markers of oxidative stress response were found to be altered including LPO, and a decrease in the levels of reduced GSH and related redox cycle enzymes such as SOD, catalase, GR and GPx was also observed(Reference Tabrez and Ahmad56). An elevated level of LPO indicates TCE-induced oxidative damage to both liver and kidneys(Reference Torasson, Clark and Dankovic57). An elevated level of MDA, a LPO product, was observed after treatment with TCE(Reference Torasson, Clark and Dankovic57, Reference Watanabe and Fukui58). In the present study, the TCE-treated rats showed a significant increase in the level of MDA, and diosmin attenuated its level in renal tissue (Table 3). Thus, diosmin exhibited the protective efficacy against TCE-induced LPO in renal tissue. TCE has been shown to inhibit the activity of antioxidant enzymes (GSH reductase, GSH S-transferase and catalase) in the renal tissue of rats(Reference Tabrez and Ahmad56). In agreement with previous studies, TCE exposure leads to a depletion in GSH content and inhibits the activities of the antioxidant enzymes GSH reductase, catalase, SOD, GPx and GSH S-transferase(Reference Ali and Sultana59, Reference Sharma, Meeran and Katiyar60). GSH is a low-molecular-weight antioxidative tripeptide, and has an essential role in the detoxification of chemotherapeutic drugs, toxicants, the metabolism of nutrients and the regulation of various pathways to maintain cellular homeostasis. Enzymatic (catalase, SOD, GR, GPx, etc.) and endogenous non-enzymatic antioxidants (GSH) were significantly restored to normal levels in the diosmin-treated groups (Tables 1–3). The diosmin treatment preserved the level of all these antioxidant enzymes as previously reported by Tanrikulu et al. (Reference Tanrikulu, Kismet and Serin Kilicoglu61) and others(Reference Dholakiya and Benzeroual26, Reference Sezer, Usta and Kocak62).
Cellular damage exhibits a good correlation with enzyme deactivation and depletion(Reference Rehman and Sultana63). Serum creatinine, LDH and BUN are sensitive markers employed in the diagnosis of TCE-induced renal damage(Reference Khan, Priyamvada and Khan10, Reference Tabrez and Ahmad56). The data obtained show profound renal damage due to TCE exposure indicative from these serum toxicity markers. The increase in the level of these enzymes in the serum and the subsequent fall in the tissue might be due to the leakage of these cytosolic enzymes (due to cell membrane disruption) into the circulatory system during TCE administration. This is indicative of the onset of renal cellular damage due to kidney dysfunction and disturbance in the biosynthesis of these enzymes, with alteration in membrane permeability. The diosmin treatment prevented TCE-induced renal toxicity, as indicated by a significant decrease in serum LDH, creatinine and BUN levels (Table 2), possibly by maintaining the renal cellular membrane integrity. This indicates a possible nephro-protective effect offered by diosmin against TCE-induced damage.
KIM-1 is a type 1 transmembrane glycoprotein that has been recommended to serve as a useful and most sensitive biomarker for renal injury in preclinical and clinical studies of drug safety evaluation and the monitoring of renal disease status(Reference Zhou, Vaidya and Brown64). The basal level of KIM-1 is very low in the kidney, but is highly increased whenever there is toxic insult to the kidneys. In the present study, KIM-1 levels were markedly increased in the TCE-treated group, thus in agreement with earlier reports(Reference Zhou, Vaidya and Brown64, Reference Ahmad, Arjumand and Seth65). However, diosmin co-administration markedly suppressed the abnormal levels of KIM-1.
Furthermore, TCE-induced oxidative stress can also cause DNA damage via causing single-strand breaks, DNA–protein cross-links and many other types of DNA base modification(Reference Clay66–Reference Tabrez and Ahmad68). The single-stranded DNA break assay detects sites on the DNA where one of the strands has been nicked: (the more the nicks, the more rapidly the DNA unbinds under alkaline conditions). The present results show diosmin as the potent inhibitor of single-stranded DNA breaks, as it effectively reduced the number of single-stranded breaks when compared with the TCE-treated group (Table 3).
Ozaki et al. (Reference Ozaki, Yamada and Nakagawara69) reported that DNA damage play an important role in the regulation of p53. The tumour suppressor gene, p53, in turn, plays a pivotal role in inducing apoptosis(Reference Chumakov70). p53 is a well-established marker for DNA damage and cell death under various conditions. Elevated expression of p53 is seen with kidney toxicity during drug and chemically induced cellular injury(Reference Lash, Hueni and Putt55, Reference Zhang, De Silva and Sun71). In the present study, TCE induced p53 expression in renal tissue in the same manner as in earlier studies(Reference Chen, Wang and Chen12), and diosmin down-regulated p53 expression in a dose-dependent manner (Fig. 2).
The Bax gene is an apoptosis-promoting member of the Bcl-2 gene family, and its up-regulation is one of the key mechanisms in programmed cell death or apoptosis. Bax protein controls cell death through its participation in the disruption of mitochondria and subsequent cytochrome c release, and is also considered to be one of the primary p53 targets. The activation of this event involves the transcription-independent and Bax-dependent release of cytochrome c with subsequent permeabilisation and disruption of mitochondrial membrane and activation of caspases(Reference Chumakov70–Reference Wang, Xiao and Ko73). Results similar to that of p53 were observed in the case of Bax expression (Fig. 3). Caspases are a family of cysteine proteases implicated in the biochemical and morphological changes that occur during apoptosis (programmed cell death). The release of cytochrome c in conjunction with other mitochondrial proteins causes further activation of caspase 9. Active caspase 9 triggers the activation of caspase 3 and other downstream events such as poly-ADP-ribose polymerase cleavage and DNA fragmentation of the apoptosis process. Thus, the above-mentioned events conclude that p53 accumulation results in the Bax-dependent release of cytochrome c, which further in concert with other cofactors and proteins up-regulates the activation of caspase-7, 9 and 3. Poly-ADP-ribose polymerase cleavage, DNA fragmentation and chromatin condensation are other events to complete the apoptotic regimen.
To assess the effect of diosmin on the activation of caspase-3, -7 and -9 induced by TCE, the caspase-3, -7 and -9 colorimetric assay (ELISA) was carried out. The results generated from the present study showed that all the caspases studied were activated in animals treated with TCE when compared with the control. This activation of caspases was in agreement with previous reports which showed activation of caspases on treatment with TCE(Reference Xu, Papanayotou and Putt74, Reference Akundi, Macho and Muñoz75). Diosmin dose-dependently decreased the level of caspases, hence strengthening our hypothesis that diosmin plays an important role in modulating apoptosis induced by TCE (Figs. 2–4).
The above-mentioned results further substantiated with the histological data that exhibited the protective effects of diosmin against TCE-induced distorted renal histo-architecture, vacuolar formation, degeneration of tubular architecture, tubular congestion, swelling and blebbing.
In conclusion, diosmin attenuates the nephrotoxicity of TCE in rats. The results provide further insight into the mechanisms of TCE-induced nephrotoxicity and confirm the antioxidant potential of diosmin. Furthermore, the findings of the present study support the role of reactive oxygen species, serum enzymes, apoptotic pathway proteins such as caspase-3, -7, -9, Bax and p53 in the pathogenesis of TCE-induced nephrotoxicity. Diosmin has a potent protective effect against the nephrotoxicity of this agent, and might be clinically useful. However, there is a need for further molecular studies in this regard before it can be taken for clinical trials.
Acknowledgements
S. S. would like to thank the University Grants Commission, Government of India, New Delhi, for providing Meritorious Research Fellowship to her student M. U. R. The contributions of the authors were as follows: M. U. R., M. T., A. Q. K., R. K., A. L., O.-O.-H. and F. A. designed and conducted the experimental work; S. S. designed the experiment and wrote the manuscript. The authors declare that they have no conflicts of interest.








