


INTRODUCTION
Hepatitis C virus (HCV) infection is one of the leading causes of death and morbidity associated with liver disease and is a major contributor to public health burdens worldwide [Reference Hanafiah, Groeger and Flaxman1, Reference Iqbal, Mcrae and Mai2]. Recent estimates showed that approximately 3–4 million people around the world are newly infected every year, and over 350 000 patients die annually due to HCV-related disorders [Reference Petruzziello3–Reference Hanafiah5]. The prevalence rate of HCV infection in China is 0·43%, and approximately 13 million people are anti-HCV positive [Reference Chen, Li and Cui6]. China is one of the most affected countries and currently has the largest number of HCV-infected people in the world [Reference Wu, Zhou and Huang7].
Patients with chronic HCV infection are the most important source of transmission. Risk factors identified for the transmission of HCV include contaminated blood products, intravenous drug use, body piercing, an infected mother at birth, sexual behaviour, dental therapy, and other unclear factors [Reference Araujo8–Reference Simmonds10]. A previous epidemiological investigation was conducted by the Department of Infectious Diseases at the Affiliated Hospital of Putian University from 2006 to 2013, revealing a high incidence (20·83%) of anti-HCV-positive patients among 3312 samples from villages along the coast of Putian. Surprisingly, the prevalence of HCV infection within these areas was extremely high compared with the average global level.
Research showed that the prevalence of HCV cases was contingent on country or region, with HCV cases representing <2% of the national population in developed countries and >15% in developing countries [Reference Hajarizadeh, Grebely and Dore11]. The prevalence of anti-HCV seroprevalence was lower than 1·5% in Asia Pacific, tropical Latin America, and North America [Reference Hanafiah, Groeger and Flaxman1]. The villages mentioned above are situated in the coastal areas of Putian in south-eastern China and are relatively impoverished and economically disadvantaged. Many patients seeking care in these villages suffer from liver problems.
In general, a high prevalence of HCV infection occurs among individuals or in areas where high-risk factors are common. However, many HCV-infected patients from the coastal rural areas of Putian were not exposed to known hazardous environments in this epidemiological investigation. In addition, we found a distinct phenomenon of familial aggregation in these areas. For example, two or even six members in some families (21%) were positive for HCV infection.
It is important to know the mechanism of transmission and pathogenesis to enable appropriate treatment and prevention of HCV infection. Indeed, the familial aggregation of HCV infection provided us with a miniature model of the pathogenesis of HCV infection. Characteristics and changes of biological genes contribute to the solution to this problem [Reference Dunford12]. Additionally, HCV exhibits enormous genetic diversity and a high degree of genetic similarity, which can be used to determine the individual HCV isolates of various subtypes [Reference Chueca, Rivadulla and Lovatti13]. Genotyping is useful for investigating infection outbreaks and for improving the general understanding of the epidemiological and virological features of HCV infection [Reference Li14, Reference Yin15]. Petruzziello et al. presented an epidemiological update of HCV genotype distribution across Europe and found a prevalence of 1·7% from 2000 to 2015. Among those cases, the frequency of genotype 1 was 55·1–70·0%, followed by genotype 2 (3·2–8·9%), genotype 3 (21·0–29·0%) and genotype 4 (4·9–5·8%) from 2000 to 2015 [Reference Petruzziello3]. They also studied the global HCV epidemiology using available data published between 2000 and 2015 from 138 countries (approximately 90% of the global population), revealing that genotypes 1 (49·1%) and 3 (17·9%) were common worldwide, followed by genotypes 4 (16·8%) and 2 (11·1%), while genotypes 5 and 6 together represented <5% [Reference Petruzziello, Marigliano and Loquercio16]. However, the precise diversity of HCV genotypes and genetic variation among patients with low-risk factors was still unknown.
The HCV genome contains a 5′ untranslatable region (5′ UTR), a highly conserved RNA structure with a single open reading frame encoding structural proteins (Core, E1, E2) and non-structural proteins (p7, NS2, NS3, NS4A, NS4B, NS5A, and NS5B), and a 3′ UTR [Reference Moradpour and Penin17]. HCV genotyping assays are currently based on the sequence of regions (5′ UTR, Core, E1, and NS5B) [Reference Yin15, Reference González-Candelas, Guiral and Carbó18]. Sequencing of an appropriate region, such as the 5′ UTR, core, E1, or NS5B, is the gold standard for discriminating HCV genotype. Furthermore, the 5′ UTR has been widely utilized for genotype distribution due to its high degree of conservation [Reference Marco19].
In this study, we combined the molecular biology techniques with genotype distribution, genetic variability, and phylogenetic analysis to further identify the genotype and genetic variation of HCV-infected patients with low-risk factors in Putian coastal regions of China. A potential genetic mechanism of transmission and pathogenesis in these individuals with a low risk of HCV infection might provide important information for future treatment and prevention of HCV infections.
MATERIALS AND METHODS
Ethics statement
Ethical approval for the study was obtained from the Affiliated Hospital of Putian University, China. Informed consent was obtained from each patient before the specimens and survey information were collected.
Subjects and inclusion criteria
Samples were collected from the coastal rural areas of Putian City in south-eastern China, in which participants had anti-HCV antibodies or tested positive for HCV RNA. The patients had never received treatment for chronic infection with HCV. The expression of HCV RNA in each patient was more than 103 copies/ml. Blood samples were gathered and serum was isolated according to conventional methods. All eligible serum samples had detectable HCV RNA obtained from patients who were anti-HCV positive according to their clinical diagnosis. Additionally, a complete and detailed epidemiological questionnaire was used to assess the risk factors for HCV infection, including age at diagnosis, gender, ALT (alanine aminotransferase) levels, educational level, history of liver disease, family medical history, blood donation/transfusion history, dental therapy and surgical intervention. Valid follow-up information for the participants was recorded between July 2014 and December 2016. Diagnostic criteria for HCV infection were based on the new prevention guide on chronic hepatitis C infection in China, 2015 [20].
Exclusion criteria
All patients were negative for HBV (hepatitis B virus), HDV (hepatitis D virus), and HIV (human immunodeficiency virus) antibodies. Patients with known alcohol abuse, drug problems, pregnancy, and Child-Pugh grade B/C were also not considered in this study. Patients who had high-risk factors, such as a medical history of selling blood, blood transfusion, receiving blood products, haemodialysis, intravenous drug use, body piercing, an infected mother at birth, high-risk sexual behaviour and dental therapy, were excluded from this study.
Verification method
Plasma was preserved at −70 °C until further use or at −20 °C for 4 h for immediate use. The samples were measured for anti-HCV antibodies using commercial third-generation enzyme immunoassays from two manufacturers (InTec Products Core Anti-HCV EIA, C 20 100 068; Kehua Core Anti-HCV EIA, C2101012061, Shanghai, China). The serum samples that reacted with both of the kits were determined to be positive for HCV infection, although autoimmunity disorders can sometimes generate false-positive results. The method used to verify the detection of HCV RNA can increase the reliability of the results. The participants were labelled according to a specific numbering system to identify their individual information.
RNA extraction and amplification
HCV RNA was extracted from the serum of subjects using the QIAmp Viral RNA Kit according to the manufacturer's instructions (Qiagen, Chadstone Centre, Vic, Australia). The extracted RNA was eluted in 50–100 µl RNase-free buffers or distilled water. Complementary DNA (cDNA) was synthesized from 5 µl of eluted RNA using the SuperScript VILO cDNA Synthesis Kit (Life Technologies, Carlsbad, CA, USA) with random hexamer primers. The reactions were heated on a thermocycler (Roche Applied Sciences, Mannheim, Germany) at 37 °C for 15 min, 50 °C for 5 min, and 95 °C for 5 min and maintained at 4 °C [Reference Lamoury, Jacka and Bartlett21].
The HCV genotype was determined via nested-polymerase chain reaction (PCR) and sequencing based on the 5′ UTR region of the genome. The primers used for the nested-PCR amplification of the fragment of the 5′ UTR region are listed in Table 1. A product of approximately 230 bp from the 5′ UTR region was amplified with a modified version of a previously described protocol [Reference Chueca, Rivadulla and Lovatti13]. Then, the 20 µl reaction mixture (containing 5 µl template cDNA) was reacted in the thermocycler. The first round to amplify a 340 bp fragment was performed for 40 cycles using the outer primers Out-F and Out-R with initial denaturation at 94 °C for 5 min, denaturation at 94 °C for 10 s, annealing at 56 °C for 20 s, and elongation at 72 °C for 1 min, followed by a final extension at 72 °C for 5 min [Reference Makvandi, Ranjbari and Makvandi22]. Then, 2 µl of PCR product was used for the second-round amplification of a 230 bp fragment in a 20 µl reaction mixture containing the inner primers In-F and In-R, and the thermal cycling parameters described above were applied. Subsequently, 3 µl of PCR product was electrophoresed on a 1% agarose gel with the DL2000 marker. The amplified products were quantified using a NanoDrop spectrophotometer.
Table 1. Outer and inner primers for nested-PCR amplification of the 5′ UTR region from HCV genomic RNA
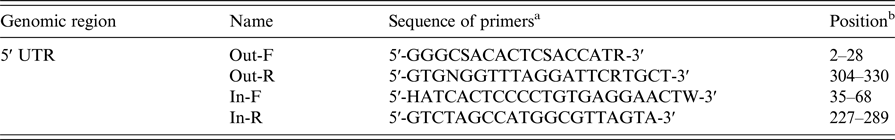
a For degenerate primers, R = A or T, W = A or T, S = G or C, K = G or T, Y = C or T, B = C or G or T, D = A or G or T, H = A or C or T, V = A or C or G, N = A or C or G or T.
b Position with reference to HCV isolate sP256432, GenBank accession KU180718.1.
Sequencing and phylogenetic analysis
The amplicons were purified using the PCR Product Purification Kit (Roche Diagnostics GmbH, Mannheim, Germany) for sequencing to identify the HCV genotype. DNA sequencing was performed by two different companies: Adicon Clinical Laboratories, Inc., and KingMed Clinical Laboratory, China. The sequencing chromatogram was visualized using Chromas v1·45chs. The assembled sequences were analysed using Lasergene v8 (DNASTAR, Madison, WI, USA). The HCV genotype was assigned using BLAST (http://blast.ncbi.nlm.nih.gov/Blast.cgi) or the HCV Sequence database (http://hcv.lanl.gov/content/sequence/HCV/ToolsOutline.html) [Reference Wasitthankasem23]. The sequences of the 5′ UTR fragment were aligned with reference sequences from the GenBank database (https://www.ncbi.nlm.nih.gov/nucleotide) using ClustalX v1·8·31. A phylogenetic tree was constructed according to the neighbour-joining method using DIVEIN (PhyML 3·0) and MEGA 4 [Reference Yin15].
Data and statistical analysis
The experiments were carried out with a minimum of two replicates. Means and standard deviations (s.d.s) were calculated and analysed for continuous variables. One-way ANOVA was used to determine the differences among the variables. The Chi-square test or Fisher's exact test was used to identify differences in the frequencies of genotype distribution and genetic variation. All statistical analyses were performed using statistics software (Stata version 12·0, StataCorp, College Station, TX). A probability (P) value <0·05 was considered to be statistically significant.
RESULTS
Epidemiological characteristics of HCV patients
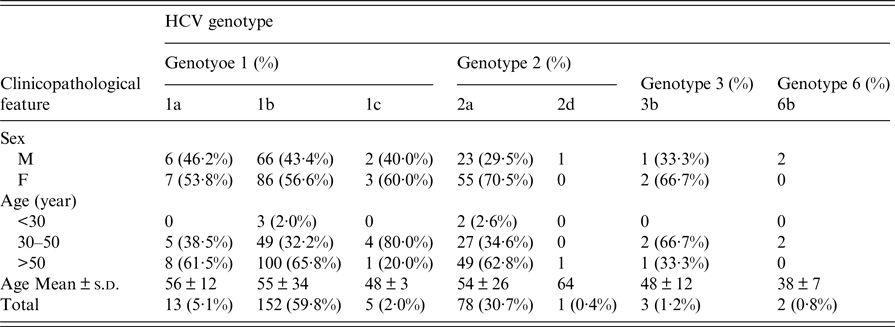
According to the criteria listed above, a total of 254 full and detailed cases that included blood serum and questionnaires were eventually included in this study. The clinicopathological and demographic characteristics of 254 HCV-infected patients from coastal rural areas of Putian City in south-eastern China are shown in Table 3. Among them, 101 (39·8%) were males and 153 (60·2%) were females. Overall, the HCV-positive patients selected for this study were 21–82 years old (mean age of 55 ± 34 years). The prevalence of HCV infection was approximately 2% in the <30 years age group, 35% in the 30–50 years age group, and 63% in the >50 years age group. People >50 years of age had a significantly greater frequency of HCV infection. Furthermore, we found that several HCV patients had infected parents, brothers or sisters, such as isolate No. 138-31469 (1b), who had a daughter with an HCV infection; isolate 180-31589 (2a), whose elder brother and sister were infected with HCV; and isolate 185-31602 (1b), who had an older sister with an HCV infection. All of the patients were of Han ethnicity.
Amplicon analysis
HCV-infected samples were further assessed for genetic variability by sequencing the 5′ UTR region, conducting a BLAST analysis, and determining the genotype distribution. The 5′ UTR region of the HCV genome (from nucleotide (nt) 2 to 340) was amplified from the total RNA of 254 HCV-positive samples collected in the villages of Putian coastal areas. The amplification of the 5′ UTR fragment was performed using nested-PCR, and the primers are shown in Table 1. A band of approximately 230 bp was detected using gel electrophoresis to confirm the size and quality of the band. Then, the fragments were independently sequenced by two companies. The electrophoresis results and chromatograms of the obtained sequences in the 5′ UTR region of HCV isolates are shown in Figures 1 and 2, respectively.

Fig. 1. Gel electrophoresis results for 5′ UTR fragments amplified by nested-PCR. M: DL2000 marker; P13: HCV isolate No. 013-30635.

Fig. 2. Chromatogram of the sequence obtained from the 5′ UTR region of HCV isolate No. 013-30635.
BLAST and sequence analysis
The 5′ UTR sequencing results from two independent companies had a high similarity of 99·8%. One of the HCV samples that were analysed in a BLAST homology search was listed after a 100% analogous identity was found. The nucleotide sequence of the 5′ UTR fragment from HCV-positive isolate No. 013-30635 was determined: TCTTGCGGGGGCACGCCCAAATGGCCGGGCATAGAGTGGGTTTATCCAAGAAAGGACCCAGTCTTCCCGGCAATTCCGGTGTACTCACCGGTTCCGCAGACCACTATGGCTCTCCCGGGAGGGGGGGGCCTGGAGGCTGTACGACACTCATACTAACGCCATGGCTAGACGCTTTCTGCGTGAAGACAGTAGTTCCTCACAGGGGAGTGA.
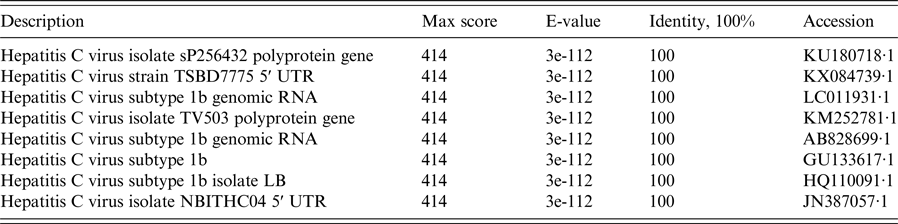
Nucleotide sequences of the 5′ UTR regions from 254 HCV RNA-positive samples were used as the input query for NCBI BLAST to find the most likely similar sequences. BLAST results revealed that highly similar sequences included identification of the 5′ UTR regions of the HCV genome. Details of the similarity of partial BLAST analysis are summarized in Table 2. The overall per cent nucleotide identity (PNI) in the 5′ UTR regions of the HCV genome was 97–100%. A partial BLAST result of the No. 002-18325 isolate with 100% of PNI and 414 of the maximum score is shown in Table 2. BLAST analysis revealed that the amplification of the 230 bp fragment was 100% homologous with the hepatitis C virus isolate sP256432 polyprotein gene (accession number KU180718·1) from nt 37 to nt 260.
Table 2. NCBI nucleotide BLAST results for the sequence in the 5′ UTR region of HCV isolate No. 002-18325 (partial alignment of the results)
The multiple sequence alignment using ClustalX software v1·8·31 for the 254 HCV RNA-positive samples compared with the GenBank database is presented in Figure 3. The nucleotide sequences of the 5′ UTR region were considered to be highly conserved. However, some mutations and variations in the 5′ UTR region of HCV gene are shown in Figure 3.

Fig. 3. Comparison of nucleotide sequences of the partial 5′ UTR region from 254 HCV RNA-positive patients and reference sequences obtained from the GenBank database. The partial multiple sequence alignment was produced by ClustalX v1·8·31 software.
Genotype distribution of the patients
Four HCV genotypes (1, 2, 3, and 6) and subtypes were detected among the 254 HCV-infected patients via sequencing analysis. The majority of the HCV-positive samples belonged to genotype 1b (59·8% 152/254), followed by genotypes 2a (30·7% 78/254), 1a (5·1% 13/254), 1c (2·0% 6/254), 6b (0·8% 3/254), and 2d (0·4% 2/254), as shown in Table 3. However, no other genotypes were detected.
The genotype distribution of HCV-positive patients is presented in Figure 4. As shown in Table 3 and Figure 4, the overall genotype ratios of HCV-positive females were higher than those of males. Samples with genotype 1a (average age of 56 ± 12 years) included six (46·2%) males and seven (53·8%) females. Of the genotype 1b patients with a mean age of 55 ± 34 years, 66 (43·4%) were males and 86 (56·6%) were females. Of the 78 patients with genotype 2a (mean age of 54 ± 26 years), 23 (29·5%) and 55 (70·5%) were males and females, respectively. There was no statistically significant difference (P > 0·05) in the average age among HCV patients infected with genotypes 1a, 1b, and 2a.
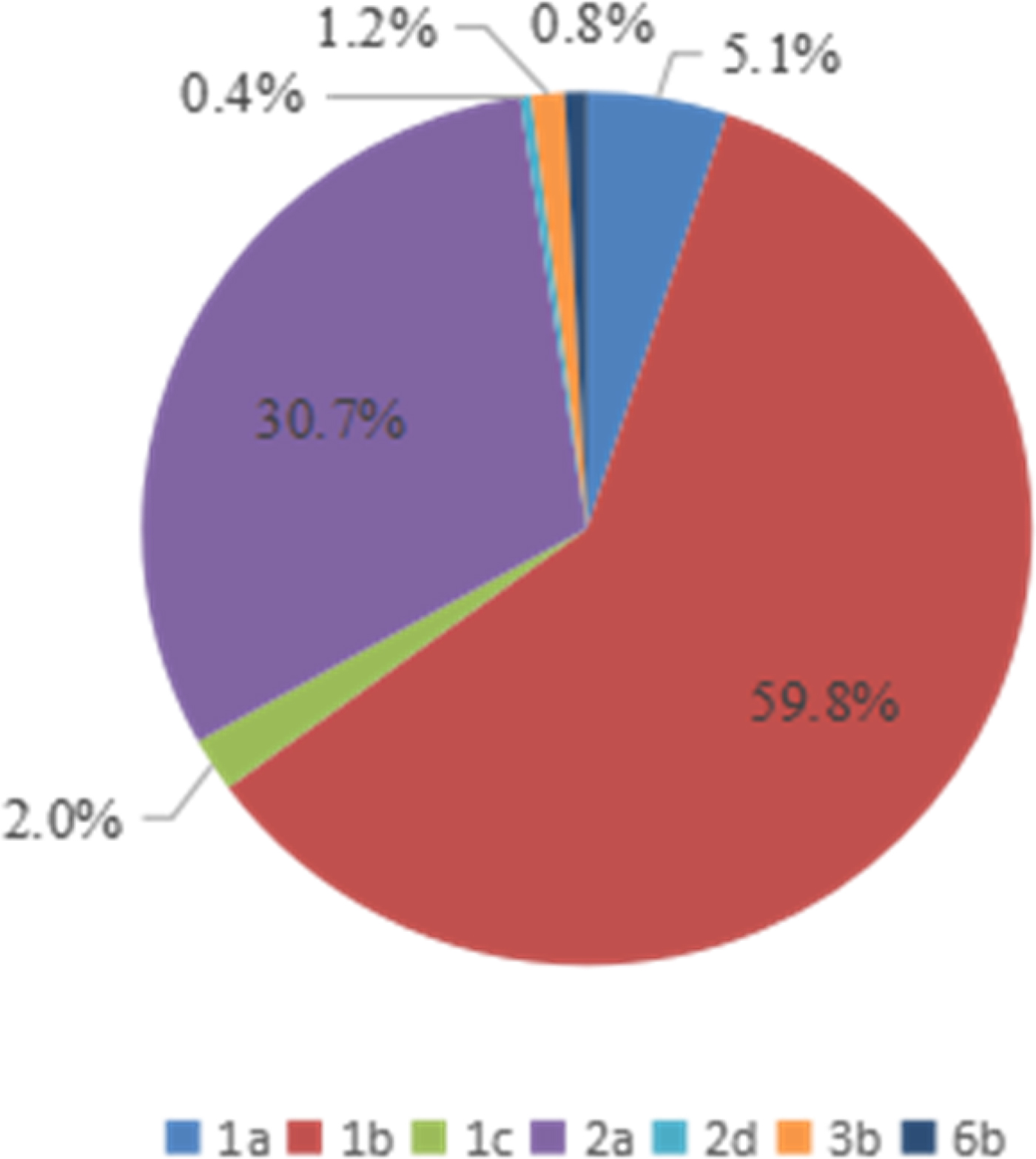
Fig. 4. Genotype distribution of 254 HCV-positive patients from the Affiliated Hospital of Putian University, China.
Table 3. Correlation between clinicopathological features and HCV genotype frequencies among 254 HCV-infected patients from the Affiliated Hospital of Putian University, China
The youngest HCV-positive patient was a 21-year-old male with genotype 1b. The oldest patient was an 82-year-old female with genotype 1b. The correlation between age and genotype (including 1a, 1b, and 2a) distribution is illustrated in Figure 5. In this study, no significant differences were observed in the age-specific prevalence of different HCV genotypes (1a, 1b, and 2a). The majority of HCV-infected patients were >50-years old (61·5% of genotype 1a, 65·8% of genotype 1b, and 62·8% of genotype 2a). Low prevalence rates for patients in the age range of 30–50 years were found, with 38·5% presenting genotype 1a, 32·2% presenting genotype 1b, and 34·6% presenting genotype 2a.
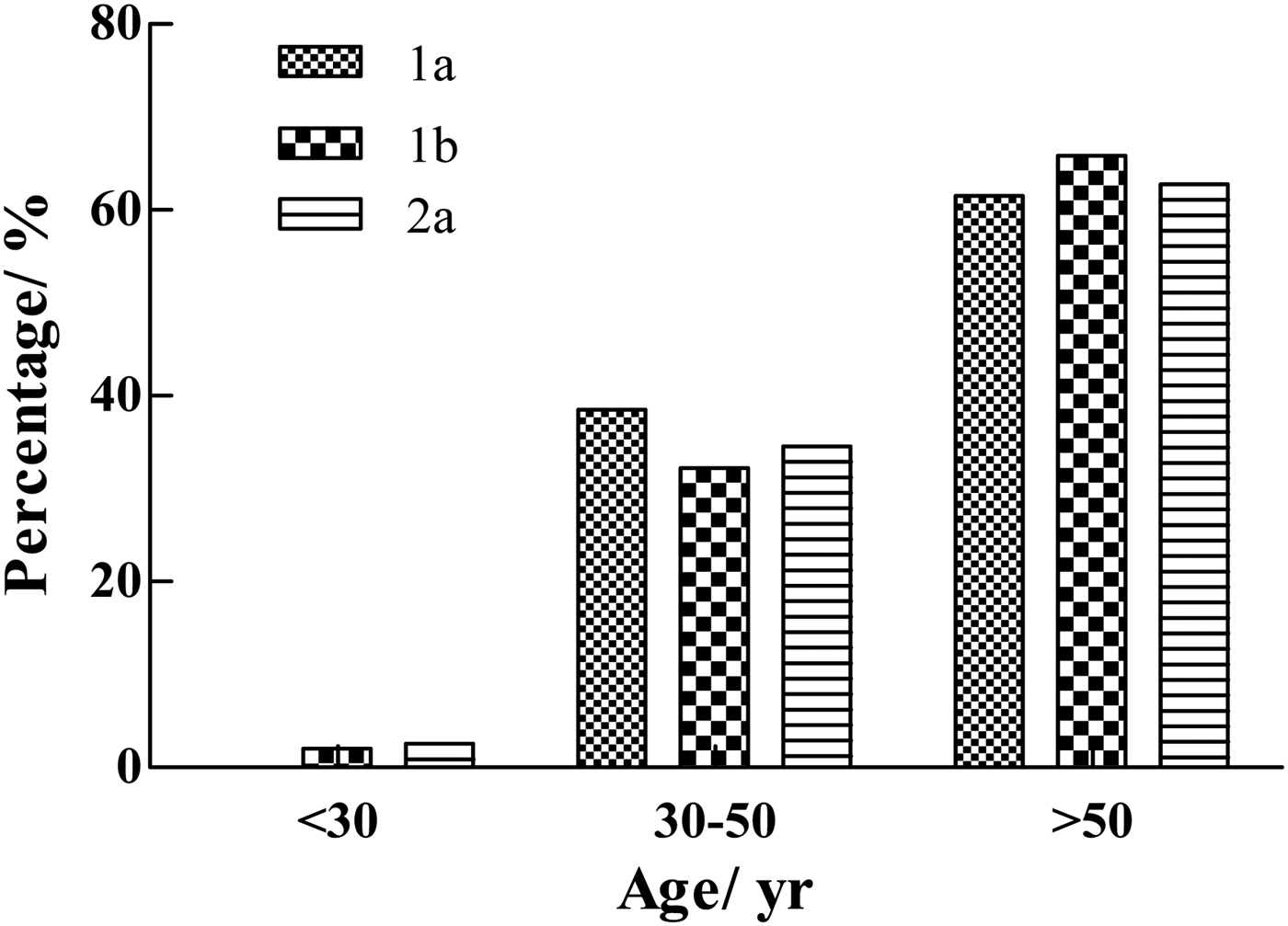
Fig. 5. Age-specific prevalence of genotypes (1a, 1b, and 2a) of HCV-positive samples from the Affiliated Hospital of Putian University, China.
Genetic mutations and variations
Sequences in the 5′ UTR region of 254 HCV infections were analysed for genetic mutations, including insertions, deletions, transitions, and transversions. High mutation rates (18·1%, 46/254) were identified in the 5′ UTR sequences, with 58·7% (27/46) of mutations found in genotype 1b and 32·6% (15/46) found in genotype 2a. Point mutations (i.e., base substitutions) were identified in 14·2% (36/254) of the sequences, and 5·1% (13/254) of specimens contained base deletions and insertions. Among these point mutations, all samples had a single point mutation in the 5′ UTR region, dual point mutations in four specimens, and three point mutations in two specimens. The majority of point mutations in the 5′ UTR region appeared ranging from nt −122 to nt −264. Single point mutations of the 5′ UTR region occurred with high frequency at nt −86 (n = 2), nt −94 (n = 2), nt −127 (n = 2), nt −138 (n = 2), nt −139 (n = 2), nt −200 (n = 2), nt −221 (n = 2), and nt −264 (n = 2). A sample showed three point mutations in the 5′ UTR region at nt −127, nt −224, and nt −226. The base insertion of HCV samples (n = 6) was found in the 5′ UTR region within a range of nt −202 to nt −237, and two of these insertions presented at the same site (nt −221). The deletions in the 5′ UTR region of HCV samples (n = 7) were discovered within a range of −83 to −294 nucleotides in length, and three of those deletions presented at the same site (nt −294). Transitions and transversions failed to be detected. Interestingly, there was a particular sample that exhibited two deletions, at nt −91 and nt −114, and one point mutation at nt −182.
Phylogenetic analysis
The high-scoring reference sequences from the GenBank database were homologous to the query sequence of the 5′ UTR region from 254 HCV-infected isolates. The phylogenetic tree was constructed with the neighbour-joining method (PhyML 3·0 and MEGA 4) using 500 bootstrap replicates for sequence alignments, as shown in Figure 6. According to the phylogenetic tree, the amplified fragments of 254 HCV RNA-positive patients were analysed along with 111 reference sequences of distinct genotypes derived from NCBI BLAST results. Phylogenetic analysis revealed that the main genotypes in 254 HCV-positive patients from the coast of Putian were genotypes 1b and 2a. Four distinct genotypes (1, 2, 3, and 6) were identified in this study. For example, genotype 1b isolate No. 009-30622 was highly homologous to the reference strain sP256432, which was reported by Simmonds and Barnes from University of Edinburgh, UK, in 2015. Comparatively, the genotype 2a isolate No. 017-30643 was strongly consistent with MRS40 at 100% homology, which was submitted by Jordier in 2012 in France. Therefore, the phylogenetic tree of HCV 5′ UTR sequences demonstrated that 152 (59·8%) strains were closely related to subtype 1b and 78 (30·7%) strains were of subtype 2a. The prevalences of other subtypes are illustrated in Figure 6 and Table 3. Furthermore, the sequences in the 5′ UTR region of HCV isolates discovered in this study will be uploaded onto the NCBI platform.
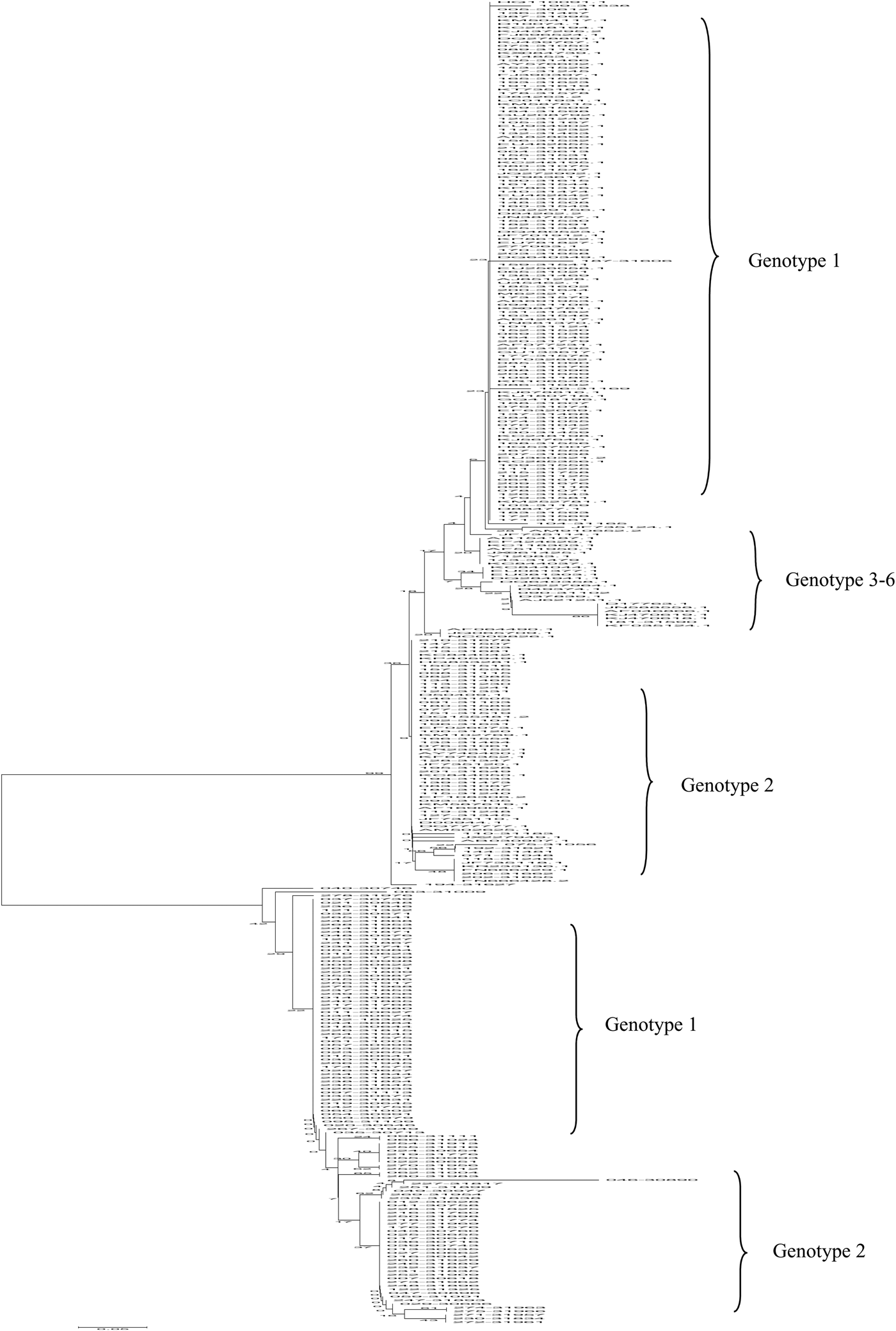
Fig. 6. Phylogenetic analysis of the 5′ UTR region from 254 HCV-infected isolates and reference sequences were generated via the neighbour-joining method using DIVEIN (PhyML 3·0) and MEGA 4 with 500 bootstrap replicates. Sequences of HCV-positive samples from the Affiliated Hospital of Putian University, China, were labelled as No. 163-31548. The reference sequences are listed by their GenBank accession number. The branch lengths were proportional to the evolutionary distance between the sequences and distance scale.
DISCUSSION
The genotypes and subtypes of HCV vary greatly in different areas, and the distributions of these genotypes have obvious geographical and population differences [Reference Nguyen and Keeffe24–Reference Patiño-Galindo, Salvatierra and González-Candelas27]. Genotyping is beneficial for analysing the route of transmission of hepatitis C and is helpful for the early detection of new subtypes of epidemic outbreaks that may have serious consequences and for providing a scientific basis for the prevention and control of HCV [28, Reference Lontok29]. With the rapid development of globalization and increasing linkage among the international community, the international population flow has become more frequent [Reference Tian30]. This phenomenon has led to the loss of geographical barriers, resulting in the rapid transmission of the HCV genotype across countries. Therefore, the regional distribution of the HCV genotype provides support for epidemiological studies.
HCV genotype 1 is found worldwide, and it is the major genotype found in the USA, Europe, Australia and Japan [Reference Marco19, Reference Zein31]. In most parts of China, the predominant HCV genotype is genotype 1b, followed by genotype 2a [Reference Lu32, Reference Xia33]. However, there are differences in genotypes among different regions. The most prominent feature of HCV in China is that there is a low prevalence rate in most areas but a high prevalence rate in some regions along with a scattered and uneven distribution of cases. Our previous epidemiological investigation found that some villages have extremely high HCV infection rates compared with the average global level. These villages are found in the coastal areas of Putian in south-eastern China, and many of these patients have low-risk factors for HCV infection. However, the exact diversity of HCV genotype and genetic variation in these patients with low-risk factors had remained unknown. In the present study, we investigated the HCV genotype by sequencing the 5′ UTR region of 254 HCV-infected positive samples with low-risk factors from the Affiliated Hospital of Putian University (China) to analyse the genotype distribution and genetic variation. According to Table 3, genotypes 1b (59·8%) and 2a (30·7%) were the most prevalent genotypes, followed by genotype 1a (5·1%). Additionally, genotypes 3b and 6b were detected in only 1·2% and 0·8% of cases, respectively. Genotypes 4, 5, and 7 were not detected in our study, which was an outcome similar to the results of Dunford et al. [Reference Dunford12]. The distribution of genotypes and subtypes varies according to geography. Our results were also consistent with the results of Yin et al., who found 36 samples (53·73%) that were subtype 1b and 31 samples (46·27%) that were subtype 2a in rural Hebei Province [Reference Yin15]. However, some differences between the present study and that of Dunford et al., who reported subtypes 1a (33%), 1b (27%), 2a (0·4%), 3a (0·7%), 3b (1·1%), 6a (18·8%), 6e (6·0%), 6 h (4·6%), and 6 l (6·4%) of HCV infections in Vietnam [Reference Dunford12]. In addition, gender distribution showed that the prevalence rates of major subtypes (1a, 1b, 1c, 2a, and 3b) of anti-HCV-positive females were higher than in males (Table 3). However, Yin et al. reported that HCV subtype 2a was predominant in females and subtype 1b was predominant in males [Reference Yin15]. This feature indicated a discrepancy in gender distribution based on country and region.
HCV infections are a major source of transmission. Risk factors identified for the transmission of HCV include transfusion of blood products, intravenous drug use (IDU), body piercing, an infected mother at birth, high-risk sexual behaviour, and other unconfirmed methods [Reference Simmonds10, Reference Alter34, Reference Nelson, Mathers, Cowie and Hagan35]. Astoundingly, the HCV-infected patients from the coastal rural areas of Putian included in this survey do not have exposure to known high-risk factors. In addition, with the implementation of medical hygiene procedures and adequate screening of blood donors over the past 10 years, the blood source and iatrogenic routes of transmission have been restricted. Nevertheless, transmission routes in areas with a high prevalence (20·83%) of anti-HCV positivity and the significant familial aggregation phenomena in the rural coastal regions of Putian remain an enigma. Contemporary studies have shown that high incidences of HCV infection have primarily occurred in undeveloped countries and regions, such as Africa and Southeast Asia. For example, the high prevalence of HCV positivity in Egypt was >10% of the general population, and this prevalence rose up to 16·8% among the blood donor population [Reference Hajarizadeh, Grebely and Dore11, Reference Awadalla, Ragab and Nassar36]. Hanafiah et al. also reported in 2005 that the prevalence of HCV infection varied depending on country and region, such as Central and East Asia (>3·5%), Australasia and Western Europe (1·5–3·5%), and North America (<1·5%) [Reference Hanafiah, Groeger and Flaxman1]. Huang et al. reported that the high prevalence (28·5%) of anti-HCV positivity was due to blood selling in the late 1980s in rural Hebei Province, North China [Reference Huang37]. The prevalence of HCV-infected individuals with low-risk factors (including voluntary blood donors and pregnant women) was only 0·5%, and the prevalence rates were significantly higher among intravenous drug users (55·6%) and dialysis patients (26·6%) from Vietnam [Reference Dunford12]. Previously, a high prevalence rate (28·86%, 303/1050) of the anti-HCV-positive cases in a village of Putian was reported by Lin et al. [Reference Lin, Huang and Liu38]. Furthermore, we speculated that the high prevalence (20·83%) of anti-HCV positivity and the significant familial aggregation phenomena might be caused by the HCV lineages inherited from ancestors. However, we did not consider vertical transmission to be an important factor because the incidence rate of HCV infection in individuals <30 years (20–30 years) was approximately only 2%, with a rate of 35% in individuals in the 30–50-year-old age group and 63% in the >50 year group, which was the main infection group. Although the patients did not present any history of blood selling, blood transfusion, drug abuse, hemodialysis, body piercing, vertical transmission, sexual behaviour or dental therapy in our study, the ancestors of these patients may have been be infected with HCV through some unknown route. In addition, we predicted that individuals might have been infected through vaccine inoculation for immunization in the 1980s when the safety and preventive treatments for iatrogenic transmission were not effective. In addition, the high prevalence of HCV infection in the coastal rural areas of Putian City may be due to the absence of effective prophylactic treatments among family members to prevent chronic HCV infection and patient-to-patient transfusion.
With respect to genetic evolution, we also analysed the genetic variation for these patients using molecular biology techniques. The results demonstrated that genetic variation was present in 18·1% (46/254) of the 5′ UTR sequences. Meanwhile, the proportion of mutation and variation in genotype 1b was 58·7% (27/46), which was more than in genotype 2a (32·6%, 15/46). Genetic variations in the 5′ UTR sequences of HCV were identified, including point mutations, deletions, and insertions. Basically, the results were consistent with most studies, and no transitions or transversions were detected. The genetic variability of the virus is an important factor for the limitation of clinical treatment. Kanwal and Mahmood identified the genetic variability and modification in the genomic regions of 5′ UTR, the core and NS5B as the underlying factors enabling the virus to escape the immune response [Reference Kanwal and Mahmood39]. González-Candelas et al. also reported that viral genetic variability might play an important role in the HCV transmission pathway, host susceptibility factors for infection or immune system response and serve as a powerful tool for tracing transmission [Reference González-Candelas, Guiral and Carbó18]. Therefore, we can speculate that the genetic diversity and variability in the 5′ UTR fragment might be the reason HCV is able to evade host defences and develop drug resistance.
There are a few limitations in this study. Relevant research methods were mainly used to analyse the gene sequence of the 5′ UTR fragment but did not include the core and NS5B gene sequences. In addition, some patients with HCV may experience repeated infections. Therefore, the ultimate reason for high prevalence of patients with low-risk factors is still unknown. We suggest that the route of HCV infection in these areas may be transmitted by lineages inherited from the ancestors. Additionally, HCV infection may have occurred via iatrogenic factors of vaccine inoculation before the 1980s, when China was relatively impoverished and economically undeveloped. Thus, a lack of adequate prophylactic treatments may be a general cause for patient-to-patient transfusion, which should be further investigated in subsequent studies.
CONCLUSION
In this study, we analysed the genotype distribution and genetic variation of HCV-infected patients with low-risk factors using molecular biology techniques. The results showed that genotype 1b was the most common, followed by genotype 2a and genotype 1a. In addition, genetic variations in the 5′ UTR sequence of HCV were identified, including point mutations, deletions, and insertions. The frequency of genetic variation in genotype 1b was higher than in genotype 2a. We suggest that the genetic variability of HCV may play an important role in immunosurveillance and clinical treatment for abnormal binding sites. Therefore, genetic variation may have an important effect on antiviral therapy for HCV. These variations may also provide an important experimental basis for the diagnosis and the treatment of liver disease caused by HCV in these coastal regions. Meanwhile, it has practical value for the development of diagnostic reagents and HCV vaccines.
ACKNOWLEDGEMENTS
This study was supported by the Science and Technology Project of Putian [2014S06(6)], the Natural Science Foundation of Fujian Province of China (2015J01506), and the Tianqing Hepatopathy Research Foundation of Hepatitis Fund Association of China (TQGB20150078). We thank all the volunteers who enrolled in our study, the clinical research staff and nurses for their assistance in recruitment. We thank Adicon Clinical Laboratories and the KingMed Clinical Laboratory for kindly providing the primers for sequencing and performing the sequencing analyses.
AUTHOR CONTRIBUTIONS
G. X. LIN conceived and designed the experiments; X. M. LI, R. X. QIU, and C. H. SONG performed the surveys; X. M. LI and C. H. SONG analysed the data; Q. H. HUANG, X. D. WANG, Z. T. HU, X. Z. HE, X. Y. YE, X. G. HUANG, and F. F. ZHENG contributed materials; X. M. LI, R. X. QIU, and C. H. SONG wrote the paper.
DECLARATION OF INTEREST
The authors declare no conflicts of interest.
INFORMED CONSENT
Written informed consent was obtained from the patients prior to study participation.









