


Folates are important cofactors in a large number of metabolic processes, such as amino acid interconversion, nucleotide synthesis and methylation. For this, different C1-forms of tetrahydrofolate are present in the cell. For example, 10-formyltetrahydrofolate is the C1-donor in de novo biosynthesis of purines. In the conversion reaction of uracil into thymidine by thymidilate synthase, 5,10-methylenetetrahydrofolate is the C1-group donorReference Zeisel1, Reference Gu, Wu, Qiu, Jennings and Li2. 5,10-Methylenetetrahydrofolate is also part of the methylation cycle, which supplies cells with S-adenosyl methionine (SAM). SAM is the universal methyl donor for a wide range of substrates, such as lipids, hormones, chromosomal DNA and proteinsReference Scott3. A shortage of folates leads to impaired activity of these metabolic processes, resulting in several diseases, possibly including colon cancerReference Li, Presnell and Gu4–Reference Stover7. Although the essential role of folate in basic cellular processes was largely established over 30 years ago and has been extended by more recent folate-depletion studiesReference Li, Presnell and Gu4, Reference Stover7, Reference Lucock8, little information is available on cellular adaptation to different extracellular steady state levels of folic acid. This information is essential to allow evaluation of effects of chronic sub-optimal and elevated folic acid levels. Elevated folic acid levels need attention, since in tumour-prone animals both folate deficiency and folic acid supplementation promote the progression of established neoplasmsReference Bashir, FitzGerald and Goodlad9–Reference Kim13.
We chose to use the HT29 colon cancer cell line; because this is a tumour-derived and commonly used human colon epithelial cell line. To achieve a steady-state condition, a 3-week exposure time was used. The cells were exposed to pteroylmonoglutamic acid, the folate used in fortified foods and supplements. Normally, serum folate levels are in the range of 3–10 ng/ml, while with supplementation values of 100 ng/ml can be obtainedReference Lucock8, Reference Rampersaud, Kauwell and Bailey14. Therefore, we exposed the cells to pteroylmonoglutamic acid at concentrations of 10 and 100 ng/ml. To ensure that vitamin B12 deficiency would not disturb the results, a normal human serum level of vitamin B12 at 500 pg/ml was used in each exposure. Our exposure conditions are physiologically relevant and contrast with most in vitro studies, which used full folate depletion and/or supraphysiological folate concentrations (4000 ng/ml)Reference Hayashi, Sohn, Stempak, Croxford and Kim15–Reference Novakovic, Stempak, Sohn and Kim18, and did not include vitamin B12.
Experimental methods
Cell culture
The human colon cancer cell line HT29 (ATCC, Manassas, VA, USA) was grown, in duplicate, on Dulbecco's modified Eagle's medium (DMEM) without folic acid (Invitrogen, Breda, The Netherlands), supplemented with pteroylmonoglutamic acid at 10 or 100 ng/ml for three passages. Vitamin B12 was added to DMEM, since this is the only micronutrient that is involved in the folate–methionine cycle which is not present in DMEM. Cells were seeded in 75 cm2 culture flasks at respectively 80 000 and 25 000 cells/cm2, to avoid growth-phase differences. Cells in both conditions were harvested near the exponential phaseReference Charrier, Jarry, Toquet, Bou-Hanna, Chedorge, Denis, Vallette and Laboisse19–Reference Viallard, Lacombe, Trocheris, Tabacik and Aliau21, and grown at 37°C in air with 5 % CO2 and 100 % relative humidity. The growth medium was supplemented with NaHCO3 (3·7 g/l; Sigma, St Louis, MO, USA), non-essential amino acids (1 × ; ICN, Zoetermeer, The Netherlands), fetal calf serum (5 %; Invitrogen; this provides a final medium concentration of 0·66 ng 5′-methyltetrahydrofolate/ml (1·2 nmol/l)), penicillin (5000 units; Sigma) streptomycin (5 mg/l; Sigma) and methylcobalamin (vitamin B12; 500 pg/ml; Sigma). Folate concentrations in the media were verified by HPLC analysis, as described below. Culture media were replaced every 2 d. Cells were split at 70–80 % confluence and harvested at 90 % confluence. Differences in cell growth were measured by re-plating 2·5 × 106 cells (10 or 100 ng PGA/ml) in 75 cm2 cell-culture flasks after being in culture for 3 weeks and determination of the amount of cells per flask after 3 d of culture (in triplicate), using the Coulter Counter (Beckman, Mijdrecht, The Netherlands). Total RNA was extracted using TRIZOL according to the supplier (Invitrogen). mRNA was isolated from total RNA by poly(A)+ selection using oligod(T) Sephadex (mRNA purification kit; Pharmacia, Roosendaal, The Netherlands). Concentrations were determined spectrophotometrically at A260nm and all samples were checked on 1 % tri(hydroxymethyl)-aminomethane-acetate-EDTA–agarose gels. Samples were stored at − 80°C for cDNA microarray analysis. From nine flasks of each exposure, the cells were trypsinised and the cell pellets were stored at − 80°C for folate, Fe, SAM and S-adenosyl homocysteine (SAH) determination. Before storage a sample was taken from each cell pellet, and used to determine the protein content (in duplicate) using the Bio-Rad Detergent Compatible protein assay (Bio-Rad, Veenendaal, The Netherlands).
Analysis of folate metabolism
5-Methyl tetrahydrofolate concentrations were measured in triplicate using the HPLC method of Konings et al. Reference Konings22. Twenty volumes (200 μl) of extraction buffer (50 mm-2-(N-cyclohexylamino)ethanesulfonic acid–N-(2-hydroxyethyl)-piperazine-N′-2-ethanesulfonic acid (CHES–HEPES; pH 7·85) with mercaptoethanol and ascorbic acid) were added to the cells. The mixture was heated for 5 min in a boiling water-bath and cooled. To each extract 50 μl of rat plasma conjugase was added and incubated for 4 h at 37°C. This mixture was treated further according to Konings et al. Reference Konings22. The size of the affinity column was adapted to the sample volume. SAM and SAH were determined in triplicate using the HPLC method of Melnyk et al. Reference Melnyk, Pogribna, Pogribny, Yi and James23. Instead of coulometric detection, UV detection at 260 nm was used. The method was adapted for cultured cells by addition of a thaw–freeze cycle for three times, at the beginning of the procedure. Cell lysis by thaw–freezing cycle was checked on a sample by microscopical observation of tryphan blue incorporation. The CV for multiple injections (n 10) of the same sample within a run was 4·7 % for SAH and 3·4 % for SAM.
Analysis of parameters for cell turnover
Reduction of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) by mitochondrial succinate dehydrogenase was used as a marker for metabolic activity (viability) of the cells (two independent replicate determinations of n 36) as described by Huveneers Oorsprong et al. Reference Huveneers Oorsprong, Hoogenboom and Kuiper24. Intracellular ATP production was determined (two independent replicate determinations of n 30), as a marker of cellular energy status, by the CellTiter-Glo Luminescent Cell Viability Assay (Promega, Leiden, The Netherlands). 5-Bromo-2′-deoxy-uridine (BrdU) incorporation into the DNA of growing cells was measured (two independent replicate determinations of n 36) as a marker of proliferation (BrdU Labelling and Detection Kit II; Boehringer Mannheim, Roche Diagnostics, Almere, The Netherlands). Caspase 3 and 7 activities were measured (two independent replicate determinations of n 36) as a marker of apoptosis (Apo-One Homogeneous Assay; Promega). After each assay, a sample was taken from every sample and used to determine the protein content by the Bio-Rad Detergent Compatible protein assay (Bio-Rad). Total protein was used to correct for differences in cell numbers between samples. E-cadherin was measured as a marker of differentiation (two independent replicate determinations of n 2). Whole-cell lysates were prepared by 5 min boiling of cell pellets with 1 ml 200 mm-sucrose, 20 mm-tri(hydroxymethyl)-aminomethane (pH 7·4), 1 mm-DTT and protease inhibitors (Roche Diagnostics). Protein concentration was determined using the Bio-Rad Detergent Compatible protein assay (Bio-Rad). Determination of actin and E-cadherin protein expression was achieved by SDS-PAGE (Criterion Precast system; Bio-Rad) and Western blotting. Proteins were transferred to Protran nitrocellulose membrane (Schleicher and Schuell, ‘s Hertogenbosch, The Netherlands) before immunodetection with antibodies directed against actin (Santa Cruz, 1:200, SC-10 731) and E-cadherin (1:200, M3612; DakoCytomation, Copenhagen, Denmark). Horseradish peroxidase-conjugated anti-goat (V8051) and anti-mouse (W4021) (Promega; 1:7500 and 1:2500) were used for the colour detection with tetramethylbenzidine-stabilised substrate for horseradish peroxidase (Promega). Densitometric determination of actin and E-cadherin bands was performed with Imagemaster 1D (Pharmacia).
Analysis of gene expression
cDNA microarray analysis (array construction, mRNA labelling and hybridisation) was performed as describedReference Pellis, Franssen-van Hal, Burema and Keijer25. Indirect labelling of mRNA was used, by incorporation of aminoallyl dUTP in the cDNA and subsequent chemical coupling to Cy5 monofunctional dye. A standard reference sample, consisting of a pool of mRNA from all exposures, was used and coupled to Cy3 monofunctional dye. Before hybridisation, the Cy3- and Cy5-labelled samples were mixed 1:1 (v/v) and hybridised to a cDNA microarray. This cDNA microarray contains 1056, duplicate, Caco-2-derived cDNA (A.A.C.M. Peijnenburg, unpublished results) and 192 control sequences and known genes selected for functional relevance in folate metabolism, folate transport, DNA repair and carcinogenesis. All cDNA were sequenced and annotated by means of BLAST searches in NCBI. Redundant clones were removed from the dataset, leaving 1026 unique annotations. Density values multiplied by the area of each spot, and the background (surrounding entire template) were collected using Arrayvision (Imaging Research, London, ON, Canada). Before data correction, background subtraction was performed. If the variation was less than 30 %, the median value was taken from the duplicate clones of the duplicate microarrays, otherwise this clone was discarded from further analysis. These values were used for principle component analysis (GeneMath, Applied Maths, St Marthens-Lathem, Belgium), to find variances between treatments.
Analysis of intracellular iron
Intracellular Fe concentration was analysed, in triplicate, using atomic absorption spectrometry (PerkinElmer SIMAA 6100, with graphite furnace and Zeeman background correction; PerkinElmer, Waltham, MA, USA). Sample preparation was done for the cell lines as follows. Millipore filtered water (0·5 ml) was added to the cell pellet and mixed and the cell suspension was sonicated for 15 min. A 10 μl sample was stored at − 20°C for protein concentration (in duplicate) using the Bio-Rad Detergent Compatible protein assay (Bio-Rad, Veenendaal, The Netherlands). Cells suspensions were digested after addition of 1·0 ml 70 % HNO3 (Baker, Philipsburg, NJ, USA) by 6 h incubation at 65°C, followed by an overnight incubation at room temperature and subsequent 6 h incubation at 65°C. Millipore filtered water (1·0 ml) was added and additionally incubated at 65°C overnight. Mixtures were sonicated for 5 min and an eleven-time dilution was prepared. Finally, the atomic absorption was measured at 248·3 nm. The CV for multiple injections (n 10) of the same sample within a run was 4·1 %.
Statistical analysis
Statistical comparisons were made using an unpaired, two-tailed Student's t test with a confidence level of 95 % using SPSS 10.0 software for windows (SPSS Inc., Chicago, IL, USA).
Results
When adapting the HT29 cells from normal culture media (4000 ng folic acid/ml) to the different folic acid concentrations, a striking difference in growth was observed during the second passage of culture. The cells grown on 100 ng folic acid/ml exhibited a 2·4-fold (P = 0·005) higher growth rate measured as cell-number increase than cells cultured on 10 ng folic acid/ml (Table 1). This was confirmed by analysis of MTT conversion, a parameter of metabolic activity that is often taken as a parameter of proliferation. A 2·1-fold (P = 0·008) higher MTT conversion was obtained at 100 ng folic acid/ml. Increased metabolic activity of cells grown at 100 ng/ml was confirmed by 6·3-fold (P = 0·0002) higher intracellular ATP levels. Cell growth was stopped when the cells were split after one passage of full depletion of folic acid (data not shown).
Table 1 Parameters of growth, folate metabolism and cell turnover*
(Mean values and standard deviations)
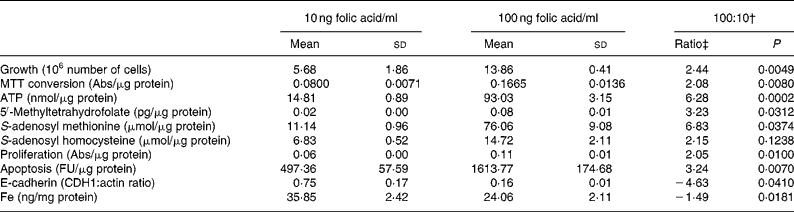
MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; Abs, absorbtion; FU, fluorescence units.
* All parameters were normalised for protein content, except growth and E-cadherin. Data were analysed by an unpaired, two-tailed Student's t test with a confidence level of 95 %.
† 100:10, -Fold change of 100 ng folic acid/ml:10 ng folic acid/ml.
‡ A negative ratio implies a higher expression at 10 ng folic acid/ml.
Analysis of folate metabolism
We determined the three most common cellular folate metabolites; 5-methyltetrahydrofolate, SAM and SAHReference Sohn, Stempak, Reid, Shirwadkar, Mason and Kim26, Reference Mason27. All three metabolites were present in higher concentrations in cells exposed to folic acid at 100 ng/ml than at 10 ng/ml (Table 1). Intracellular 5-methyltetrahydrofolate levels, a measure for intracellular folate status, increased 3·2-fold (P = 0·03), SAM levels increased 6·8-fold (P = 0·04) and SAH levels increased 2·2-fold (P = 0·12).
Analysis of gene expression
To gain more insight in the long-term effect of folic acid exposure we used a relatively small cDNA microarray representing 2200 genes, to investigate genes involved in folate metabolism, colorectal cancer and the cell cycle. Expression of ten genes was higher in HT29 at the 100 ng folic acid/ml condition (Table 2), the expression of six genes was found to be higher at folic acid exposure of 10 ng/ml. Strikingly, none of these genes is related to folate metabolism. The expression of three tumour-related genes was higher at folic acid exposure of 100 ng/ml. Nine genes involved to cell-cycle processes were affected. More specifically, the expression of four genes engaged in increasing proliferation was up regulated in the 100 ng folic acid/ml condition, while the expression of two genes that up regulate apoptosis was also higher. In total, four genes that are involved in down regulation of differentiation were found for the 100 ng/ml compared with the 10 ng folic acid/ml condition. Surprisingly, the expression of two genes linked to Fe metabolism was found to be decreased at folic acid exposure of 100 ng/ml. Below, we further assessed the effects on cell turnover and Fe metabolism.
Table 2 Description of the differentially expressed genes
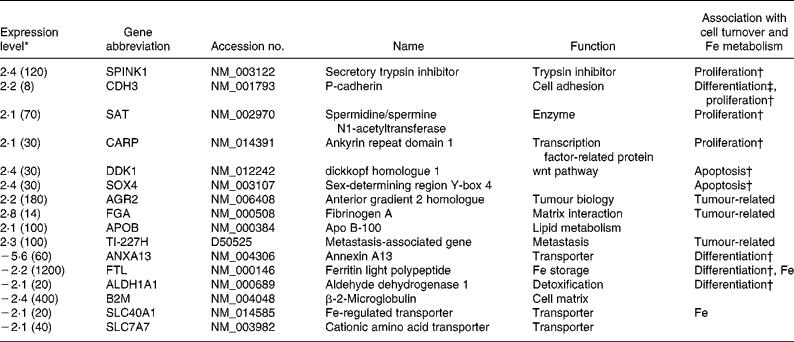
* The 100 ng folic acid/ml:10 ng/ml ratio is given; ratios greater than 2 are indicated. A negative ratio implies a higher expression at 10 ng folic acid/ml. The values in parentheses indicate expression levels and are shown as signal divided by 10 times the background.
† Up regulation of this gene is associated with up regulation of this process.
‡ Up regulation of this gene is associated with down regulation of this process.
Analysis of parameters for cell turnover
To verify the observed differences in cell turnover we used different physiological assays and Western blot analysis rather than quantitative real-time RT-PCR (Q-PCR), since the responses measured using Q-PCR are not necessarily of physiological relevanceReference Valasek and Repa28. Cell turnover is a resultant of proliferation, apoptosis and differentiation; often in folate research only one cell turnover parameter is examinedReference Gu, Wu, Qiu, Jennings and Li2, Reference Li, Presnell and Gu4, Reference Huang, Ho, Lin, Wei and Liu29, Reference Singh, Fouladi-Nashta, Halliday, Barrett and Sinclair30. The cells grown on 100 ng folic acid/ml had 2-fold (P = 0·01) higher proliferation and also a 3·2-fold (P = 0·007) higher rate of apoptosis. Differentiation was analysed by determination of E-cadherin protein expression using immunoblotting. In the 100 ng folic acid/ml exposure a 4·6-fold (P = 0·04) lower E-cadherin protein level was found as compared with the 10 ng folic acid/ml exposure (Table 1).
Analysis of intracellular iron
To confirm the effect of folic acid on Fe metabolism we determined intracellular Fe concentrations. A 1·5-fold (P = 0·02) lower intracellular Fe level was found in the 100 ng folic acid/ml as compared with 10 ng folic acid/ml exposure. A lowering of ferritin protein was also detected (data not shown).
Discussion
A chronic exposure of folic acid at 100 ng/ml compared with a 10 ng/ml exposure resulted in higher concentrations of intracellular 5′-methyltetrahydrofolate, of SAM and of SAH, in higher cell growth and in lower differentiation and in lower intracellular Fe levels in the HT29 colon cancer cell line.
Only a small number of genes were affected. This is in agreement with other microarray-based papers that have investigated the effect of folic acid on gene expression. The papers that studied cell lines all compared folate depletion and supraphysiological folate exposure. Although extreme differences in folate concentration were used, also under these conditions only a relative small number of genes (3·40 %) responded, except for the study of Novakovic et al. Reference Novakovic, Stempak, Sohn and Kim18 in which 5–24 % response was found. Remarkably among the differentially expressed genes we did not find any gene involved in folic acid metabolism. In agreement with this, Crott et al. Reference Crott, Choi, Ordovas, Ditelberg and Mason31, using colonic rat mucosa, and Jhaveri et al. Reference Jhaveri, Wagner and Trepel17, using nasopharyngeal epidermoid carcinoma cells, also found that folate depletion did not affect the expression of any folate-related genes. This seems in contrast with Courtemanche et al. Reference Courtemanche, Huang, Elson-Schwab, Kerry, Ng and Ames16, who used non-epithelial cells. In primary human T lymphocytes these authors found that folate deficiency resulted in down regulation of genes involved in folate metabolism, in particular the folate transporter. The use of a different cell type may explain the differences between the studies. This is supported by a recent study which specifically examined the expression of genes involved in folate metabolism in the colon cancer cell lines Caco-2 and HCT116 and showed an effect of folate deficiency on folate metabolism gene expression, with clear differences between the two cell linesReference Hayashi, Sohn, Stempak, Croxford and Kim15. Besides the use of different cell types, a possible explanation for this difference could be the length of exposure and the different cut-offs used in gene selection. Courtemanche et al. Reference Courtemanche, Huang, Elson-Schwab, Kerry, Ng and Ames16 also reported higher expression of DNA repair, as was true for Novakovic et al. Reference Novakovic, Stempak, Sohn and Kim18, and mitochondrial genes. The effect on mitochondrial genes points to an alteration of cellular energy metabolism. This agrees with physiological observations in the present study, which showed an effect of folate on ATP content and MTT conversion. Mitochondrial folate metabolism is of physiological relevance, since one-carbon units are required for the synthesis of formate, glycine and f-met-tRNA in mitochondria. Furthermore, mitochondria play an important role in carcinogenesisReference Augenlicht, Bordonaro, Heerdt, Mariadason and Velcich32, Reference Penta, Johnson, Wachsman and Copeland33. Due to the limited size of our cDNA microarray we cannot gain insight into the processes lying underneath. The precise role of folate on mitochondrial physiology and cellular energy metabolism is not well investigated, which is an area that deserves further attention.
Cell proliferation and apoptosis are essential features in gut renewal, but excess cell proliferation is associated with carcinogenesis and generally considered as one of the early events in colon cancerReference Bashir, FitzGerald and Goodlad9, Reference Arends34. Recently, animal studies have indicated that timing and dose of folate intervention may be of critical importance in protection from colon carcinogenesisReference Bashir, FitzGerald and Goodlad9–Reference Kim11, Reference Kim13. It was observed that folate supplementation seems to enhance established neoplasms and deficiency has an inhibitory effectReference Bashir, FitzGerald and Goodlad9–Reference Kim11. This is in contrast to normal epithelium, in which folate deficiency appears to increase neoplasm risk, whereas moderate folate supplementation suppresses the development of tumoursReference Li, Presnell and Gu4, Reference Le Leu, Young and McIntosh35–Reference Hirohashi39. The observed higher growth rate was accompanied by a higher rate of both proliferation and apoptosis. This implies that the rate of cell turnover, and therefore cell growth, is determined by the balance between proliferation and apoptosis, and that analysis of only one of these parameters is not a good measure for the ultimate physiological effect. A key feature in early and later stages in colon cancer is the inactivation of the differentiation marker E-cadherin proteinReference Hirohashi39–Reference Buda and Pignatelli41. Disappearance of E-cadherin protein expression is indicative of increased colon cancer risk. Here, we showed that exposure to a higher level of folic acid resulted in a lower level of E-cadherin protein expression. Increased differentiation was also found in methotrexate-treated HT29 cellsReference Singh, Fouladi-Nashta, Halliday, Barrett and Sinclair30 and increased cadherin-associated protein-β-catenin 1 gene expression in folate-depleted HT29 cellsReference Novakovic, Stempak, Sohn and Kim18. Together with our findings of enhanced cell growth and reduced differentiation, results are supportive of the suggestion that the dose of folic acid may be critical in protection of carcinogenesis or otherwise enhancement of cancersReference Bashir, FitzGerald and Goodlad9, Reference Kim10, Reference Wright, Finglas and Southon42.
We found that intracellular Fe levels are lower at exposure to folic acid at 100 ng/ml, which shows an effect of folate on Fe metabolism. We also found a lowering of total Fe by the 100 ng folic acid/ml exposure in the human colon cancer cell line Caco-2 and in the SV-40 immortalised human colon cell line CCD841CoTr (data not shown). This is most probably not due to kinetic differences between the 10 ng/ml and 100 ng/ml folic acid-exposed cells, since DMEM has an excess of Fe. Previously, the converse, an effect of Fe status on folate levels has been established. It was shown that Fe deficiency resulted in folate deficiency in animals and human subjectsReference Westerman, Evans and Metz43–Reference Vitale, Streiff and Hellerstein45. Also a direct interaction of Fe on folate metabolism has been shown; reduction of free Fe by chelation of Fe by either the chemical chelator mimosineReference Oppenheim, Nasrallah, Mastri and Stover46 or the biological chelator ferritin heavy chain (but not ferritin light chain)Reference Oppenheim, Adelman, Liu and Stover44 resulted in up regulation of the translation rate of cytoplasmic serine hydroxyl methyl transferase (cSHMT) and increased folate catabolism. We did not find an effect of folate levels on cSHMT mRNA expression (data not shown), but we cannot exclude an effect on SHMT protein levels. Our observation, that folate affects total cellular Fe and the expression of ferritin light chain indicates a close interaction between both micronutrients. This interaction may have a function in the regulation of cellular single-carbon metabolism. This warrants further research, not only with respect to colon cancer risk, but also in view of the important role of Fe and folate for the developing fetusReference Picciano47.
Acknowledgements
We thank the members of Food Bioactives group of RIKILT-Institute of Food Safety for discussions. We thank Dr Peter Hollman for advice on folate and SAM/SAH analysis and Dr Ad Peijenburg for help with the development of the Caco-2 cDNA microarray. The present study was financially supported by grant WS 99–72 from the MLDS, the Dutch Digestive Diseases Foundation, by grant VCZ 980–10–020 from ZonMw, the Netherlands Organization for Health Research and Development and by grant 803–71–54 801 from the Ministry of Agriculture, Nature Management and Food Quality.


