


INTRODUCTION
Benign epilepsy of childhood with central temporal spikes (BECTS) is the most common benign partial epilepsy of childhood, accounting for 13%-23% of all childhood epilepsies.Reference Cavazzuti1, Reference Kriz and Gazdik2 Typical EEG findings consist of diphasic spikes or sharp waves with prominent after-coming slow waves. Spikes have a characteristic of horizontal dipole configuration, with maximal negativity in the centro-temporal regions and positivity in the frontal regions.Reference Gregory and Wong3, Reference Gregory and Wong4 It is well established that this EEG pattern can occur in patients without clinical seizures.Reference Cavazzuti, Cappella and Nalin5 Conversely, childhood absence epilepsy (CAE) and juvenile absence epilepsy (JAE) are the different forms of generalized epilepsies, accounting for 2%-10%Reference Jallon and Latour6 and 0.2%-3%Reference Jallon and Latour6–Reference Loiseau8 of childhood epilepsies, respectively. Characteristic EEG findings include synchronous and symmetrical spike-wave paroxysms of at least 3 Hz associated with brief impairment of consciousness.Reference Dravet9, Reference Wolf10
Although both EEG findings have age-dependent occurrence, absence of significant anatomic lesion on neuroimaging, multifactorial etiology, genetic predisposition and favorable long-term outcome, they are distinct electro-clinical entities that rarely overlap in the same individual. Some suggest that the conditions may be part of a continuum,Reference Beaumanoir, Ballis, Varfis and Ansari11 while others contend that their co-existence is coincidental.Reference Gelisse, Genton and Bureau12, Reference Cerminara, Coniglio, El-Malhany, Casarelli and Curatolo13 We describe 20 patients with the co-existence of 3 Hz generalized spike-wave (GSW) and rolandic discharge on EEG over a period of 25 years. Three had BECTS and later developed absence epilepsy, whereas 17 had absence epilepsy with rolandic discharges on EEG, without typical rolandic seizures. This small fraction of patients with co-existence of both EEG findings is suggestive that they are distinct electro-clinical markers that likely do not share a common etiology.
METHODS
All EEG investigations at BC Children’s Hospital (BCCH) are entered in a database, which included EEG findings and clinical data obtained by an EEG technologist at each visit. The database was queried for patients with both 3 Hz GSW discharges and benign rolandic discharges (centro-temporal spikes and sharp waves with centro-temporal negativity and frontal positivity) on simultaneous or separate EEGs between 1992 and 2017.
Clinical data, including demographics, seizure types and frequency, underlying and/or co-existing neurological diagnosis, medications, development and neuroimaging were obtained from the database and a retrospective review of medical records was conducted. Seizure type was classified according to 2017 International League Against Epilepsy as CAE, JAE or BECTS.Reference Fisher, Cross and French14 Clinical outcome was evaluated by reviewing developmental milestones, neurologic exam, seizure frequency and medications at presentation and follow-up.
RESULTS
Among the 43,061 patients in the EEG database from 1992 to 2017, 1426 patients were identified with benign rolandic discharges and 528 patients with 3 Hz GSW discharges on EEG, and 20 patients (0.05%) were found to have both EEG findings on the same or follow-up EEGs (Tables 1 and 2).
Table 1 Features of patients with BECTS who later developed CAE. Clinical features of patients with BECTS and absence epilepsy

AED = anti-epileptic drug; CLB = clobazam; DD = developmental delay; F = female; M = male; VPA = valproic acid
Table 2 Features of patients with CAE who also had rolandic trait on EEG. Clinical features of patients with absence epilepsy and benign rolandic trait

AED = anti-epileptic drug; CLB = clobazam; DD = developmental delay; ETX = ethosuximide; F = Female; LAM = lamotrigine; LEV = levetiracetam; M = Male; PHB = phenobarbital; VPA = valproic acid.
Patients with BECTS Who Developed CAE
Of the 20 patients, three fulfilled the diagnostic criteria for BECTS and CAE (Table 1). In these patients, as BECTS resolved, they developed CAE. Age of onset of seizure ranged from 4 to 9 years of age. All patients presented with the characteristic seizure manifestation of BECTS, including symptoms of hemi-facial or hemi-body motor involvement, mainly affecting the face and orthopharynx with speech arrest and hyper-salivation accompanied by a typical EEG pattern with sharp waves in the centro-temporal regions with characteristic of horizontal dipole and normal background. One had a family history of epilepsy and one had a family history of febrile seizures. Two had a history of febrile seizures before the onset of BECTS. Two patients had neuroimaging (MRI), which were normal. The conversion of one seizure type to another was not secondary to medications. Patient 1 had BECTS until 13 years of age and was not treated with any anti-seizure medications. He developed CAE at 11, with an overlap of the seizure types for 2 years. Patient 2 had BECTS from 4 to 9 years of age and treated with valproic acid (VPA) and subsequently clobazam (CLB), which was weaned at 11. At 13 years of age, she developed absence epilepsy. Patient 3 was diagnosed with BECTS at 6 years of age and treated with CLB. After being seizure-free for 3 years, this medication was weaned. As she was weaning the medication, she developed CAE, which did not resolve with the re-introduction of CLB, but to ethosuximide (ETX). All patients had long-term follow-up, ranging from 9 to 12 years. Patients 1 and 2 were seizure-free at last follow-up and Patient 3 had rare seizures. One had a mood and anxiety disorder and another had severe anxiety. All three patients had normal intellectual functioning.
We provide a more detailed clinical description of an exemplary patient (Patient 3 in Table 1).
Patient 3
At 6 years of age, a right-handed female was referred to the neurology clinic for a 3-minute seizure consisting of staring, speech arrest, rhythmic mouth movements, excessive drooling and hand fumbling, with relative preservation of consciousness. Electrocardiogram showed central temporal spikes and sharp waves with characteristic horizontal dipole. She was started on CLB and had no further seizures for 3 years and the medication was weaned at age 10. MRI brain, development and neurological examination were normal. She had a history of three febrile seizures from 2 to 3 years of age. There was no family history of seizures.
At the time of the discontinuation of CLB, it was noted that she started to have absence seizures, and EEG at the time confirmed absence epilepsy, with six clinical seizures captured associated with 3 Hz GSW discharges. She continued to have left central temporal spikes and sharp waves. Clobazam was re-initiated, but the absence seizures were not controlled. Subsequently, she was started on ETX and then lamotrigine (LAM). At last follow-up, she was seizure-free on LAM, developmentally normal, but had an anxiety disorder.
Patients with Absence Epilepsy and Rolandic Trait on EEG
In addition, 17 patients were identified with absence epilepsy with 3 Hz GSW discharges and simultaneous rolandic trait, without having rolandic seizures (Table 2). Clinical and electroencephalographic features of the 17 patients were as follows: age at onset of seizures ranged from 1.5 to 7 years, with a mean of 4.5 years. All had the following ictal manifestations: abrupt loss of consciousness and quick recovery. Some had automatisms, fluttering of the eyelids and brief myoclonic jerks. All had bilateral synchronous symmetrical discharges of GSW at 3 cycles per second. Neurologic examination was normal in all and 12 had neuroimaging (11 with MRI and 1 with CT), of which 10 were normal, 1 showed non-specific delayed myelination. There was a family history of epilepsy in seven patients and family history of febrile seizures in one individual. The total number of medications each patient tried ranged from 0 to 10, with a mean of 2.53. Follow-up varied from 0.5 to 16.5 years, with a mean of 7.35 years. At the time of last follow-up 15 were seizure-free, one diagnosed with GLUT-1 deficiency continued to have daily absence seizures and one had occasional eyelid fluttering, but was not on any medications at last follow-up. Various co-morbidities were described, including attention deficit hyperactive disorder (ADHD) (four patients), anxiety (two patients), requirement of school assistance (11 patients) and developmental delay (nine patients).
DISCUSSION
We have described 20 patients with both 3 Hz GSW discharges and benign rolandic trait, representing 0.05% of patients in our EEG population. BC Children’s Hospital is the only pediatric tertiary care center in British Columbia, Canada, serving a population of 4.7 million people. There are few reports of overlap of both EEG findings in the literature (Table 3).
Table 3 Literature review: patients with history of rolandic trait and 3 Hz spike-wave discharges on simultaneous or subsequent EEGs
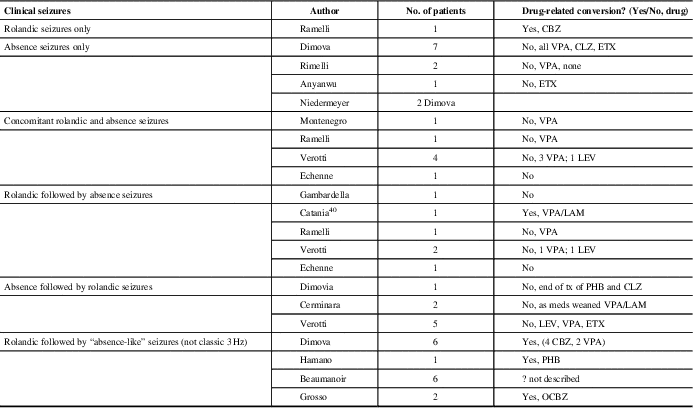
CBZ = carbamazepine; CLZ = clonazepam; ETX = ethosuximide; LAM = lamotrigine; LEV = levetiracetam; OCBZ: oxcarbamazepine; PHB = phenobarbital; VPA = valproic acid
Of our patients three were affected by BECTS, and upon remission of this idiopathic generalized epileptic syndrome with typical absences developed. Moreover, five patients with similar presentation, not thought to be drug-induced, have been previously described.Reference Gambardella, Aguglia, Guerrini, Morelli, Zappia and Quattrone15–Reference Echenne, Rivier, Roubertie, Touzery and Humbertclaude18 Gelisse et al reviewed patients over 11 years with rolandic EEG discharges. Of 66 patients identified with typical rolandic spikes, none of the patients had 3 Hz GSW. Therefore, they concluded that there is no continuum between the two EEG findings.Reference Gelisse, Genton and Bureau12 Others have described the conversion of benign rolandic to “absence-like epilepsy” that is likely drug-related.Reference Beaumanoir, Ballis, Varfis and Ansari11, Reference Dimova and Daskalov19–Reference Grosso, Balestri and Di Bartolo21 Lamotrigine, phenobarbital (PHB) and carbamazepine (CBZ) have been implicated. None of our patients were on these medications at the time of onset of the absence epilepsy.
Dimova described six patients who developed “absence-like” seizures after a history of BECTS, of which four initially received CBZ, which is known to precipitate or aggravate absence seizures in epileptic patients. In their study, none of the children demonstrated the classic 3 Hz GSW discharge with their absence seizures and there was a strong laterality of the discharges, suggesting that the absences might be of a secondarily generalized origin, unlike that observed in idiopathic absence epilepsy.Reference Dimova and Daskalov19
The transition from focal seizures to apparently generalized seizures is not a common phenomenon. There are several descriptions of “atypical partial benign epilepsy of childhood” or “pseudo-Lennox syndrome.”Reference Aicardi and Chevrie22, Reference Hahn, Pistohl, Neubauer and Stephani23 In this condition, children with focal epilepsy experience frequent atonic, atypical absence and myoclonic seizures, often with non-convulsive status epilepticus, as well as cognitive and behavioral disturbances. Some cases have been provoked by CBZ and other anti-seizure medications.Reference Guerrini, Belmonte and Genton24
Although 3 Hz GSW discharges are rare, brief subclinical bursts of bilateral abnormalities occur in about 25% of patients with BECTS.Reference Gelisse, Genton and Bureau12 Therefore, generalized GSW in BECTS has not been considered a specific marker for the continuum between focal and generalized epilepsies, as their appearance is likely the result of bilateral synchrony and not of generalized hyper-excitability.
In our study and in the literature, incidental rolandic spikes in patients with absence epilepsy were the most common finding in those with both types of EEG findings. We had 17 such patients and an additional 12 are collectively reported in the literature.Reference Ramelli, Donati, Moser and Vassella16, Reference Dimova and Daskalov19, Reference Anyanwu, Ghavami, Schuelein and Motamedi25, Reference Niedermeyer26 In general, typical rolandic discharges are seen in 0.7% of awake recordings or normal children without a history of seizures;Reference Cavazzuti, Cappella and Nalin5 if recordings had sleep, this number may be higher. The percentage of children with rolandic discharge who develop clinically apparent seizures is unclear. The risk is probably <10%, on the basis of reported incidences of BECTS and of rolandic EEG discharges in normal children.Reference Wirrell, Camfield and Camfield27 Therefore, rolandic discharges are considered most likely an incidental finding in children with seizures or spells whose semiology is not suggestive of BECTS. Benign rolandic trait could simply be a genetic expression and its overlap with CAE could be coincidental expression of two genetic predispositions.
Some have researched circuit mechanisms common to GSW discharges and focal discharges. One study demonstrated that the thalamic circuit, integral for 3 Hz generalized discharges, is capable of producing local recurrent activity that is highly organized and repetitive, such as rolandic discharges. Huntsman et al demonstrated that mutant mice that were devoid of a particular GABA, subunit (p3) lacked functional connectivity between TRN cells, which promote or desynchronize thalamocortical oscillations. These mice had absence seizures and widespread hypersynchrony in isolated thalamic slices. Of note, a small fraction (10%) of isolated thalamic slices from control animals demonstrated focal reverberant activity. Therefore, network responses that resembled the activity of absence seizures could evoke restricted activity in small regions of the thalamic slice, as seen in focal epilepsy.Reference Huntsman, Porcello, Homanics, DeLorey and Huguenard28 Another study demonstrated that rolandic discharges in sleep have a high positive correlation with delta and spindle activity.Reference Nobili, Baglietto and Beelke29 These suggest rolandic discharges may have a connection to thalamocortical activity, which is necessary for spindles and 3 Hz spike-wave discharges. Further evidence shows that the generation of 3 Hz GSW discharge relies on dual activation of the thalamus and cortex.Reference Aghakhani, Bagshaw and Benar30, Reference Gotman, Kobayashi, Bagshaw, Bénar and Dubeau31
Benign epilepsy of childhood with central temporal spikes and absence epilepsies have multifactorial etiology, with some genetic predilection, although unique from each other. As of yet, no overlapping genes have been described for both types of epilepsies. Rare cases associated with Elongator Protein Complex 4, which has roles in transcription and tRNA modificationReference Strug, Clarke and Chiang32 and KCNQ2 and KCNQ3 mutationsReference Neubauer, Waldegger and Heinzinger33 have been described. In absence epilepsy, rare monogenetic etiology has been reported. Subunits of ion channels, GABRG2, CLCN2, GABRA1 and CACNA1H have been implicated.Reference Lu and Wang34 Genetic studies in the patients with dual findings would be of great interest, to determine any link.
There are eight reported clinical cases with idiopathic generalized epilepsy onset: CAE or JAE, who, after a period of seizure freedom, experienced partial seizures with the electro-clinical characteristics of BECTS.Reference Cerminara, Coniglio, El-Malhany, Casarelli and Curatolo13, Reference Verrotti, Casciato and Spalice17, Reference Dimova and Daskalov19 This was a common presentation described by Verotti in his multicenter trial of those with both seizure types.Reference Verrotti, Casciato and Spalice17 We found no such cases in our population. There are seven cases in the literature of patients who simultaneously have both seizure types.Reference Ramelli, Donati, Moser and Vassella16–Reference Echenne, Rivier, Roubertie, Touzery and Humbertclaude18, Reference Montenegro and Guerreiro35 Again, this was not the cases in our patients; as the first seizure type was remitting, the second seizure type developed. Patient 1 (Table 1) did have an overlap of both seizure types for 2 years.
Patients with GLUT-1 deficiency have a defect in the facilitative glucose transporter GLUT-1Reference De Vivo, Trifiletti, Jacobson, Ronen, Behmand and Harik37 and may present with focalReference Wolking, Becker and Bast36 and/or generalized seizures in early infancy, including early-onset absence seizures (<4 years of age).Reference De Vivo, Trifiletti, Jacobson, Ronen, Behmand and Harik37, Reference Klepper, Willemsen and Verrips38 Multifocal or generalized discharges, including 3 Hz GSW discharges, often with background slowing, can be seen on EEG.Reference Wolking, Becker and Bast36, Reference Klepper, Willemsen and Verrips38 Early diagnosis is important, as GLUT-1 patients do not respond to anti-seizure medications, but to the ketogenic diet.Reference Klepper, Scheffer and Leiendecker39 In our study, six patients presented with absence seizures under the age of 4 (Table 2). In 5/6 patients, good response to anti-seizure medications was not consistent with GLUT-1 deficiency. One patient was tested for GLUT-1 deficiency due to presentation at 1.5 years of age and the diagnosis was confirmed by cerebral spinal fluid profile. They were subsequently treated with the ketogenic diet. This patient had no history of focal seizure semiology and rolandic spikes had characteristic horizontal dipole formation.
Overall, despite having both EEG findings, the majority of our patients had good clinical outcome, regardless of clinical presentation or number of medications needed to control the seizures. At last follow-up, most patients were seizure-free or had rare seizures. This is similar to the patients reported by Verotti, where all patients with both clinical seizure types showed excellent clinical prognosis.Reference Verrotti, Casciato and Spalice17
To the best of our knowledge, this is the largest cohort of patients in one center with the co-existence of both EEG findings in the same individual that are not drug-induced. The rarity of the co-existence of these two EEG findings implicates a separate pathophysiology and genetic susceptibility. Further multicenter and genetic studies would contribute to our understanding. Ultimately, the majority of patients, regardless of clinical presentation, showed an improvement or normalization in their EEGs of both focal and generalized paroxysmal activity and had good seizure outcome, suggesting that the presence of dual findings does not alter the favorable prognosis of either type of epilepsy.
Acknowledgments
The authors thank Jeffrey Zhi for his help in accessing archived EEGs and for his assistance with data entry. Also, thanks to Ruth Janke for helping to identify patients from the EEG database.
Financial Support
This research received no specific grant from any funding agency in the public, or from commercial or not-for-profit sectors. All work was done at BCCH.
Disclosures
AND, LW, and PKHW have no conflicts of interest to declare.
Statement of Authorship
AND developed the original concept and study design of the manuscript. She obtained clinical data by performing a detailed chart review and entering the data in a spreadsheet. She did a literature review on the subject and created Table 3. She helped to analyze the data, drafted the manuscript, then reviewed and edited it for important intellectual content.
LW participated in identifying eligible patients. She helped obtain pertinent clinical information from the EEG database. She also helped to analyze the data. She prepared Tables 1 and 2, summarizing the clinical data. She reviewed and edited the manuscript for important intellectual content.
PKHW created the EEG database, which was crucial to identify patients. He provided guidance in study design and analysis. He reviewed and edited the manuscript for important intellectual content.
All authors gave approval to the final version of the manuscript to be submitted and are in agreement to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Ethics Approval
Approved by University of British Columbia Ethics Board H17-01949.



