


The fragile X premutation (PM) consists of a small unstable CGG repeat expansion ranging from 55 to 200 repeats in the noncoding region of the fragile X mental retardation 1 (FMR1) gene (Hagerman & Hagerman, Reference Hagerman and Hagerman2004). The most severe disorder associated with PM alleles is a late-onset, progressive neurodegeneration termed fragile X-associated tremor/ataxia syndrome (FXTAS), affecting about 75% of male carriers after the age of 80 years but less than 20% of female carriers in the same age group (Hagerman & Hagerman, Reference Hagerman and Hagerman2013; Jacquemont et al., Reference Jacquemont, Hagerman, Leehey, Hall, Levine, Brunberg, Zhang, Jardini, Gane and Harris2004; Loesch & Hagerman, Reference Loesch, Hagerman and Hannan2011). The much lower risk of FXTAS in female than in male carriers can be, at least partly, attributed to the mitigating effect of the normal FMR1 allele on the second female X chromosome (Jacquemont et al., Reference Jacquemont, Hagerman, Leehey, Hall, Levine, Brunberg, Zhang, Jardini, Gane and Harris2004).
The traditional diagnostic (major) features of FXTAS required for ‘definitive’ diagnosis comprised postural and/or intention tremor, usually in the upper extremities; gait ataxia and T2 hyperintensities in the middle cerebellar peduncles on MRI (MCP sign; Jacquemont et al., Reference Jacquemont, Hagerman, Leehey, Grigsby, Zhang, Brunberg, Greco, Des Portes, Jardini, Levine, Berry-Kravis, Brown, Schaeffer, Kissel, Tassone and Hagerman2003). Other features include parkinsonism, cognitive decline, neuropathy and other white matter hyperintensities on MRI (Hagerman & Hagerman, Reference Hagerman and Hagerman2013; Loesch & Hagerman, Reference Loesch, Hagerman and Hannan2011). The revised diagnostic criteria for FXTAS additionally included T2 splenial hyperintensities as a major neuroimaging feature, and neuropathy as a minor diagnostic criterion (Hall et al., Reference Hall, Birch, Anheim, Jønch, Pintado, O’Keefe, Trollor, Stebbins, Hagerman, Fahn, Berry-Kravis and Leehey2014). These new recommendations also include broadening the range of the CGG repeat to incorporate gray zone (GZ) alleles of 41–54 CGGs, which have been shown to play a role in the risk for Parkinson’s disease (PD; Hall et al., Reference Hall, Berry-Kravis, Zhang, Tassone, Spector, Zerbe, Hagerman, Ouyang and Leehey2011; Loesch et al., Reference Loesch, Khaniani, Slater, Rubio, Bui, Kotschet, D’Souza, Venn, Kalitsis, Choo, Burgess, Johnson, Evans and Horne2009; Loesch et al., Reference Loesch, Tassone, Lo, Slater, Hills, Bui, Silburn and Mellick2013; Loesch et al., Reference Loesch, Tassone, Mellick, Horne, Rubio, Bui, Francis and Storey2018), and apparently in the etiology of typical (syndromic; Hall & O’Keefe, Reference Hall and O’Keefe2012), or atypical (Debrey et al., Reference Debrey, Leehey, Klepitskaya, Filley, Shah, Kluger, Berry-Kravis, Spector, Tassone and Hall2016) FXTAS.
The mechanisms leading to the severe neuropathological changes underlying FXTAS are complex and not fully understood (Hagerman & Hagerman, Reference Hagerman and Hagerman2013, Reference Hagerman and Hagerman2016); this may contribute to a large diversity of type and severity of clinical manifestations extending beyond the established diagnostic criteria (FXTAS spectrum; Hall & O’Keefe, Reference Hall and O’Keefe2012; Loesch et al., Reference Loesch, Churchyard, Brotchie, Marot and Tassone2005; Loesch & Hagerman, Reference Loesch, Hagerman and Hannan2011). In this study, we investigate an instance where this diversity presents as a predominant phenotype of kinetic tremor in a sibship of three males and three females, all carrying FMR1 PM or GZ alleles (see case descriptions in the supplementary material). This monosymptomatic presentation was reminiscent, but not diagnostic, of FXTAS, and was very similar in all six siblings regardless of sex and CGG repeat expansion size. We tested a hypothesis of the existence of background genetic modifiers influencing the observed phenotype, using a genomewide association study and nonparametric linkage.
Methods
Source of Sibling Series
All the participants have been involved in a major fragile X research project, ongoing at La Trobe University from 2012, and supported by the National Institutes of Health, USA. The sibling series included in this study was ascertained through a granddaughter of one of the female participants, who suffered developmental delay. Identification of the FMR1 full mutation as the cause of the delay led to cascade testing, which revealed that her mother and grandmother, as well as her grandmother’s five siblings, were all carriers of small CGG expansions. The relevant data from a cohort of six male and six female patients classified as FXTAS and investigated within the same major project were included for comparison with the siblings’ neurological profiles. All participants were aged >50 years when examined and were of Anglo-Celtic extraction. All participants provided informed consent according to protocols approved by the La Trobe University Human Research Ethics Committee (HEC01-85).
Protocols
Neurological and cognitive measures
Three motor scales — the Unified Parkinson’s Disease Rating Scale Part III-Motor (UPDRS-III; Fahn et al., Reference Fahn, Elton, Fahn, Marsden, Calne and Goldstein1987), the International Cooperative Ataxia Rating Scale (ICARS; Trouillas et al., Reference Trouillas, Takayanagi, Hallett, Currier, Subramony, Wessel, Bryer, Diener, Massaquoi, Gomez, Coutinho, Ben Hamida, Campanella, Filla, Schut, Timann, Honnorat, Nighoghossian and Manyam1997), and the Clinical Rating Scale for Tremor (CRST; Fahn et al., Reference Fahn, Tolosa, Marin, Jankovic and Tolosa1993) — were administered by two neurologists with experience of these scales.
General cognitive functioning was assessed using Addenbrooke’s Cognitive Examination Test (ACE 3; Mioshi et al., Reference Mioshi, Dawson, Mitchell, Arnold and Hodges2006). The Vocabulary and Matrix Reasoning subtests of the Wechsler Adult Intelligence Scale (Third Edition; WAIS-III) were used to calculate a prorated Full-Scale IQ score, with Matrix Reasoning also providing a measure of nonverbal reasoning. (The Similarities subtest replaced Matrix Reasoning to calculate a prorated IQ score in Case 1 in Table 1.) WAIS-III Digit Spans (but with forward and backward separately) were employed as measures of attention and working memory, respectively.
Executive functioning was also assessed using the Trail Making Test (TMT; Reitan & Wolfson, Reference Reitan and Wolfson1985) as a measure of divided attention/set shifting. The Symbol Digit Modalities Test was used as a measure of information processing speed (Smith, Reference Smith1982). The delayed recall and discrimination indices of the Hopkins Verbal Learning Test-Revised (HVLT-R; Brandt, Reference Brandt1991) were employed as measures of delayed recall and recognition memory, respectively.
Psychiatric pathology test scores
The SCL-90-R (Derogatis, Reference Derogatis1994), a 90-item self-administered questionnaire, was chosen as it can efficiently provide information on a broad range of relevant psychological symptom clusters. Here, we report a summary score providing a measure of overall psychological distress — the Global Severity Index (GSI), as well as three specific symptom domains selected a priori: Depression, Anxiety and Obsessive-Compulsive.
FMR1 molecular measures
CGG sizing: Genomic DNA was isolated from peripheral blood lymphocytes using standard methods (Purygene Kit; Gentra Inc., Minneapolis, MN). For Southern blot analysis, 10 micrograms of isolated DNA were digested with EcoRI and NruI. Hybridization was performed using the specific FMR1 genomic dig-labeled StB12.3 probe as previously described (Tassone et al., Reference Tassone, Pan, Amiri, Taylor and Hagerman2008). Genomic DNA was also amplified by PCR (Filipovic-Sadic et al., Reference Filipovic-Sadic, Sah, Chen, Krosting, Sekinger, Zhang, Hagerman, Stenzel, Hadd, Latham and Tassone2010). DNA analysis was performed in the Laboratory of Dr Tassone at the MIND Institute, UC Davis. The activation ratio: The activation ratio (AR) indicates the proportion of cells that carry the normal allele on the active X chromosome, so that AR = 1.00 indicates a normal allele active in 100% of the cells. It was measured based on the intensity of the appropriate bands on Southern blots as per published protocol (Tassone et al., Reference Tassone, Hagerman, Iklé, Dyer, Lampe, Willemsen, Oostra and Taylor1999).
Genomewide Array Genotyping and Associated Analyses
All six siblings underwent single-nucleotide polymorphism (SNP) genotyping using the Illumina Global Screening Array. This contains ~650,000 SNPs spanning the entire human genome, selected to give high-quality imputation of ungenotyped variants, especially in populations of European descent. It incorporates nearly 50,000 coding variants enriched for relevance to disease (via ClinVar). Nonparametric linkage analysis was carried out using the Allegro 2.0f linkage package (Gudbjartsson et al., Reference Gudbjartsson, Thorvaldsson, Kong, Gunnarsson and Ingolfsdottir2005). We used the affecteds-only exponential pairs LOD score analysis. With the PRSice2 package (Choi & O’Reilly, Reference Choi and O’Reilly2019), we calculated a PD polygenic risk score (PRS) for the siblings based on 107 SNPs reported to be associated with PD in Supplementary Table S2 of Nalls et al. (Reference Nalls, Blauwendraat, Vallerga, Heilbron, Bandres-Ciga, Chang, Tan, Kia, Noyce, Xue, Bras, Young, von Coelln, Simón-Sánchez, Schulte, Sharma, Krohn, Pihlstrøm, Siitonen and Zhang2019). We imputed the key SNP genotypes, when not present on the GSA, using Beagle Version 5.1 (Browning et al., Reference Browning, Zhou and Browning2018). We compared the values of this statistic in the siblings to that in a collection of 45,000 Australian controls genotyped on several different arrays, including the GSA, collected for multiple other studies at the QIMR Berghofer Medical Research Institute and in all members of the UK Biobank. Unfortunately, the number of known essential tremor (ET) genes is far lower than for PD. We did carry out genomewide association analyses looking for genetic modifiers using the same SNPs, but obviously statistical power is low for this approach, given there are only six related cases. We did perform a candidate gene association subanalysis, using previously reported ET variants in CTNNA3, STK32B, PPARGC1A, DRD3, LINGO1, and FUS (Diez-Fairen et al., Reference Diez-Fairen, Bandres-Ciga, Houle, Nalls, Girard, Dion, Blauwendraat, Singleton, Rouleau and Pastor2019; Müller et al., Reference Müller, Girard, Hopfner, Merner, Bourassa, Lorenz, Clark, Tittmann, Soto-Ortolaza, Klebe, Hallett, Schneider, Hodgkinson, Lieb, Wszolek, Pendziwiat, Lorenzo-Betancor, Poewe, Ortega-Cubero and Rouleau2016).
Results
Family History
The results of the motor, cognitive and psychiatric pathology scale scores in the sibship are listed in Table 1. Cascade testing revealed that all the proband grandmother’s (Case 6) siblings were also carriers of small CGG expansions: two brothers (Case 1 and 2), and one sister (Case 5) were PM carriers, and one brother (Case 3) and one sister (Case 4) were GZ carriers. The siblings’ mother, an obligate carrier, could not be assessed because she died at the age of 90 years, before this study. She is said to have suffered from hypothyroidism and anxiety for many years, and in her mid-80s developed a progressive tremor of unspecified type, and significant memory problems. However, we were able to assess the proband’s grandmother and all her siblings.
Table 1. Motor, cognitive and psychiatric scale individual scores in six siblings included in this presentation
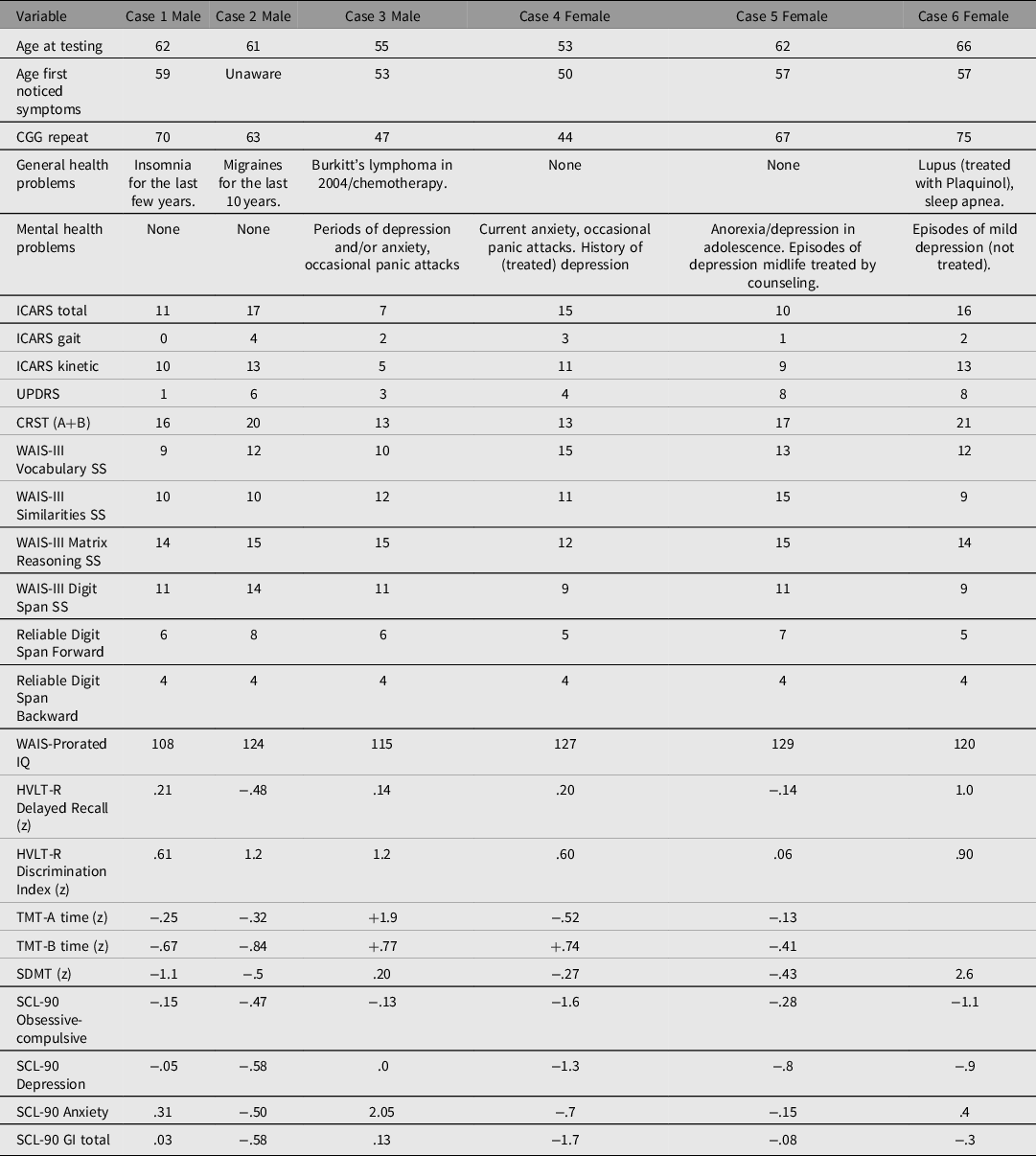
Note: +positive z scores are above average (we have inverted the z scores where necessary, so as to make a positive z scores indicative of better performance on specific tasks), and less symptomatic on SCL-90 domains.
Clinical History
All six siblings presented a broadly similar clinical picture of onset in late middle age of bilateral (though often asymmetric) kinetic (action/intention) tremor of the upper limbs. In all but one, there was slight bilateral postural tremor as well, but this was always less prominent than the action/intention tremor. Five had lower limb kinetic tremor as well; this was the predominant feature in only one female. There was no resting tremor. Excluding one male who denied a history of tremor but who was nevertheless obviously affected on examination, the mean age of symptom onset was 55.8 years (range 53−59) and the mean duration of symptoms at the time of examination was 6.4 years (range 2−13). Variations on this tremor description were the addition of ‘postural’ (held note) vocal tremor in three and postural tongue tremor in two (with one overlap); and apparent orthostatic tremor on inspection and palpation of the thighs bilaterally in one sibling. There were slight parkinsonian features (bradykinesia) in three out of six siblings.
None displayed cognitive impairment on testing. Only one sibling, a female, scored in the moderate range for depression, or for overall psychological distress, on selected SCL-90 subscales. None of the siblings reported alcohol or drug abuse.
None of the five siblings who underwent MRI studies manifested the typical, relatively specific MRI features of FXTAS, or, except for one female (GZ) carrier (Case 4), any other minor changes to an extent beyond that attributable to age.
Comparison With Motor Dysfunction Profiles of FXTAS Patients
The differences in motor dysfunction profiles between this sibship and a comparison group of six males and six females with FXTAS group-matched for age are illustrated in Figure 1. The positioning of the scores clearly shows that the severity of motor changes on the ICARS or UPDRS is milder in the siblings than in the FXTAS cohort. This especially applies to stance and gait as assessed on the ICARS, where there is minimal overlap between individual plots for siblings and comparison group. In contrast, there is obvious overlap in ICARS kinetic scores, and also, predictably, in the CRST, where the scores represent all forms of tremor. These CRST scores are much more variable in the comparison group than in the siblings. Similarly, while there is still some overlap between the two categories with respect to parkinsonian features as measured by the UPDRS scores, these features are much less prominent in the siblings than in our comparison group. Another important conclusion that can be derived from this summary plot is that while the scores on all the three scales are, predictably, much higher in FXTAS males than females, this is not the case for the siblings, where the male scores are not obviously different from the female scores.

Fig. 1. Individual plots representing ICARS, UPDRS and CRST (A+B) scale scores for sibship and FXTAS cohorts of male and female carriers.
Note: % scores are in relation to the total for the respective scales.
Genomic Studies
Considering the close clinical similarity of the siblings’ phenotype to a form of dominantly inherited ET, we have investigated potential genetic influences that might either be causal, or act as modifiers of ET or of parkinsonism to which it not infrequently evolves, throughout the entire genome.
Nonparametric multipoint linkage analysis determined that the only chromosomal region shared homozygously by all six siblings identical by descent was the short arm of chromosome 10 (10p13-10p11.22, 13.8-32.2 Mbp hg19 map). The peak exponential pairs LOD score was 3.01, which does not reach the formal threshold for genomewide significance of 3.3 (Figure 2). It is, however, the maximal LOD score achievable given the family and obviously implies an autosomal recessive model. This region does not include the reported chromosome 10 ET locus CTNN3 (at 68–69.9 Mbp). The known PD SNP rs896435 in ITGA8 does fall within the suggestive interval, and all six siblings were homozygous for the T risk allele (compared to control frequencies, p = .11). This is not a high penetrance gene, in that T/T homozygote risk is only 16% higher than baseline.

Fig. 2. Genomewide nonparametric linkage analysis for members of the affected sibship. Panel A shows the peak region on chromosome 10p, while Panel B shows the entire genome.
Moreover, we have calculated PD PRSs, based on the top 107 SNPs reported by Nalls et al. (Reference Nalls, Blauwendraat, Vallerga, Heilbron, Bandres-Ciga, Chang, Tan, Kia, Noyce, Xue, Bras, Young, von Coelln, Simón-Sánchez, Schulte, Sharma, Krohn, Pihlstrøm, Siitonen and Zhang2019) in our six siblings against these scores for UK Biobank PD cases, UK unaffected controls and Australian population controls (Figure 3). The PRSs are presented in this figure as the equivalent (genetic) predicted risk of PD by the age of 65 years based on the UK Biobank participants. We found that all the six cases fell in the top 30% of individuals in the general population for polygenic risk of PD. This does not differ significantly from the mean risk in the population (p = .7). We did not observe any genomewide significant (i.e., p < 5 × 10−8) individual SNP association results for our PD and ET candidate genes (see Supplementary Figure 1).

Fig. 3. PRS distributions: UK Biobank PD cases (red curve) and unaffected controls (black curve), Australian population controls (blue curve), and current sibship (black impulses). The median PRS for the current sibship lies at the top 32nd percentile of the control distributions.
Discussion
The separation between the PM (56−200) and GZ (41/45−54) ranges of FMR1 expansions was originally made on the basis of whether the expansion can result in a full mutation (>200) leading to fragile X syndrome in grandchildren — that is, within two generations. However, our data have shown that these two smaller expansion types may otherwise be on a continuum with respect to neurological consequences. Here, we describe a sibship of three males and three females, all of whom had potentially pathogenic FMR1-CGG expansions, either within the PM or GZ range. Given the siblings’ carrier status, the main diagnostic consideration was of a FXTAS spectrum disorder, but the contrast of motor scale scores obtained from the sibship with those from the comparison probable or definite (syndromic) FXTAS groups allowed us to detect important differences in dysfunction profiles between these two cohorts. The predominant clinical manifestations in all six siblings comprised mild to moderate postural and kinetic (action and intention) tremor, with disproportionately minor or absent stance and gait ataxia. This motor dysfunction profile is consistent with absence of the MCP sign. Three siblings also displayed mild parkinsonian features. General cognition, as well as several aspects of executive function, were intact, in contrast to FXTAS, where executive dysfunction may reflect disruption of the prefronto-ponto-cerebello-thalamo-prefrontal loops. Considering the relatively young age of the siblings, there is a possibility that this unusual monosymptomatic manifestation of the FXTAS spectrum could potentially evolve into a full syndromic form of FXTAS involving typical cerebellar changes. Toward this end, we retested Case 2 after 3 years’ interval (information given in his case description in Table 1 refers to this repeat testing) but recorded only progression in the kinetic tremor score without any additional manifestations.
In addition to the siblings’ atypical clinical profile, there was also no evidence for an X-linkage ameliorating effect in female carriers, who showed marked similarity to their male siblings in motor scores and cognitive profiles, consistent with the lack of correspondence between AR results and clinical manifestations. Neither was there any obvious effect of FMR1 allele category — PM versus GZ — on the siblings’ clinical status.
These observations suggested that other (autosomal) genetic factor(s) additive to, or interacting with, the FMR1 small expansion allele might be involved in a modified phenotypic expression consistent across the sibship and reminiscent of another diagnosable movement disorder such as ET. Indeed, recently reviewed evidence indicates that genetic modifier effects might be responsible for potential symptom overlap between various neurodegenerative diseases (Tarakad & Jankovic, Reference Tarakad and Jankovic2019). Such overlaps in cognitive and motor features between FXTAS spectrum and several other neurodegenerative disorders, including ET, have been apparent from the recent phenotypic comparisons (Robertson et al., Reference Robertson, Hall, McAsey and O’Keefe2016). The neurological profile observed in all siblings presented here was, indeed, reminiscent of ET, the most common hyperkinetic movement disorder(s) characterized by isolated postural and kinetic tremor of hands, arms, head, voice and, less commonly, legs (LaRoia & Louis, Reference LaRoia and Louis2011). The spectrum of ET may also include additional neurological signs of uncertain significance, including mild gait imbalance or parkinsonism (Bhatia et al., Reference Bhatia, Bain, Bajaj, Elble, Hallett, Louis, Raethjen, Stamelou, Testa and Deuschl2018; LaRoia & Louis, Reference LaRoia and Louis2011; Tarakad & Jankovic, Reference Tarakad and Jankovic2019). Given that ET is heterogeneous with respect to etiology and pathological mechanisms (Zhang et al., Reference Zhang, Zhao, Zhou, Li, Yi, Guo, Yan, Tang and Sun2017), and the high degree of clinical variability of both ET and FXTAS, the difficulties in differential diagnosis and in understanding the nature of commonalities between these two conditions are not surprising (Robertson et al., Reference Robertson, Hall, McAsey and O’Keefe2016).
Considering the presence of the ET-like phenotype in all the siblings, we initially performed a candidate gene association subanalysis of genomewide screening, using previously reported ET variants in CTNNA3, STK32B, PPARGC1A, DRD3, LINGO1, and FUS (Diez-Fairen et al., Reference Diez-Fairen, Bandres-Ciga, Houle, Nalls, Girard, Dion, Blauwendraat, Singleton, Rouleau and Pastor2019; Müller et al., Reference Müller, Girard, Hopfner, Merner, Bourassa, Lorenz, Clark, Tittmann, Soto-Ortolaza, Klebe, Hallett, Schneider, Hodgkinson, Lieb, Wszolek, Pendziwiat, Lorenzo-Betancor, Poewe, Ortega-Cubero and Rouleau2016), with negative results. We are aware, however, that the limitations due to the low statistical power in our small sample of only six related cases, combined with the limited number of known ET genes, cannot allow for ultimate exclusion of any potential effect of the presumed ET genotype. On the other hand, the assumption that the tremor seen in this family is solely due to ET genes is very unlikely. Even though the incidence of ET is up to 5% by the age of 70 years, and heritability reaches 90% (Clark & Louis, Reference Clark and Louis2015), the occurrence of ET in all six siblings would be extremely unlikely (only 6 in 10,000 of such sibships under the multifactorial threshold model). However, our data revealed borderline linkage of PD SNP rs896435 in ITGA8 to a broad region of the short arm of chromosome 10, with all six siblings being homozygous for the T risk allele, though these findings do not implicate any obvious candidates. The position of the siblings’ average PD PRS was in the top 30% of the Australian population distribution of these scores. We are aware that these results, although they may be suggestive of the existence of background PD risk genotype potentially modifying the FXTAS spectrum phenotype, have been obtained in a single set of siblings. It is therefore recommended that, following our initial exploration, the hypothesis of potential genetic modifiers, especially the PD risk factors, should be tested in future studies based on large cohorts in order to establish whether this type of analysis extrapolated onto the much larger population of small CGG expansion carriers on the one hand and/or of ET patients on the other can lead to more conclusive results. Especially considering that the postulated modifying effect of this parkinsonian genome, this is more likely to only apply to subpopulations of individuals in respective diagnostic categories who also manifest parkinsonian features in addition to their specific FXTAS or ET phenotypes. This assumption is substantiated by the common (in ~30%) occurrence of parkinsonian features in FXTAS, combined with the lack of ameliorating effect of the normal FMR1-X chromosome on the parkinsonian (UPDRS) score in a cohort of the female PM carriers (Loesch et al., Reference Loesch, Tassone, Atkinson, Stimpson, Trost, Pountney and Storey2021). On the other hand, the reported excess of carriers of GZ alleles in several independent PD cohorts (Hall et al., Reference Hall, Berry-Kravis, Zhang, Tassone, Spector, Zerbe, Hagerman, Ouyang and Leehey2011; Loesch et al., Reference Loesch, Khaniani, Slater, Rubio, Bui, Kotschet, D’Souza, Venn, Kalitsis, Choo, Burgess, Johnson, Evans and Horne2009; Loesch et al., Reference Loesch, Tassone, Lo, Slater, Hills, Bui, Silburn and Mellick2013; Loesch et al., Reference Loesch, Tassone, Mellick, Horne, Rubio, Bui, Francis and Storey2018) indicates a possible combined effect of these alleles and the assumed PD risk genotype. Apart from the relevance of PD to the PM/GZ phenotypic pathologies, there is ample evidence of ET–PD relationships at the clinical, pathological and genetic levels (Tarakad & Jankovic, Reference Tarakad and Jankovic2019), which may be reflected in the combination of an ET-like phenotype and the increased PD risk genotype observed in the siblings carrying small FMR1-CGG expansions.
Although our report cannot provide an unequivocal answer to this diagnostic dilemma, it contributes new observations pertaining to the poorly understood and appreciated issue of the potential role of modifying genetic influences on clinical manifestations of neurodegenerative disorders such as FXTAS. Our data also present an example relevant to the concept of the ‘FXTAS spectrum’, which is still insufficiently understood or appreciated. At the genetic level, our family data provide further support for the role of intermediate size (GZ) FMR1 alleles in motor dysfunctions.
Apart from an unusual phenotype prompting our exploration of the background genotype of the siblings, two unusual genetic features of this family relevant to FMR1 carrier status are of particular interest. First, all six siblings carried an FMR1 expansion; and second, the sibship encompasses both GZ and PM alleles. One possible explanation for these findings would be compound heterozygosity for two allele categories (GZ and PM) in the deceased mother, especially as the chance of occurrence of the carrier status in all siblings in such a large sibship from a mother carrying a single allele is low (1 in 64). However, the wide range of CGG repeat sizes (from 44 to 75) does suggest two other possibilities: expansion from a maternal GZ and contraction from a maternal PM allele. Of the two, the first option is extremely unlikely considering that GZ expansion to PM size appears to be a fairly rare event, and we could not find evidence in the literature to support such an occurrence (Nolin et al., Reference Nolin, Sah, Glicksman, Sherman, Allen, Berry-Kravis, Tassone, Yrigollen, Cronister, Jodah, Ersalesi, Dobkin, Brown, Shroff, Latham and Hadd2013; Villate et al., Reference Villate, Ibarluzea, Maortua, de la Hoz, Rodriguez-Revenga, Izquierdo-Álvarez and Tejada2020). Moreover, should the mother indeed have carried a GZ expansion (presumably of 44 repeats), further expansions to a PM size of up to 75 CGGs would have had to occur on four separate occasions to explain the repeat sizes we found, making this an even more unattractive explanation. On the other hand, contractions from maternal PM alleles to GZ or normal size categories have been shown to be not infrequent, and even multiple contractions from the same mother have been reported (Nolin et al., Reference Nolin, Glicksman, Tortora, Allen, Macpherson, Mila, Vianna-Morgante, Sherman, Dobkin, Latham and Hadd2019). For these reasons, we suggest that the mother may have carried a PM expansion, which contracted into the GZ in two of the siblings.
Supplementary material
To view supplementary material for this article, please visit https://doi.org/10.1017/thg.2021.10.
Acknowledgments
The authors wish to thank all the participants and their families for their long-term commitment to this study. The authors wish to acknowledge Eleanor Hammersley for conducting two cognitive assessments. UKB data has been accessed as part of UKBB project 25331 ‘Gene mapping and genetic correlation across complex traits’.
Author contributions
1. Research project: A. Conception, B. Organization, C. Execution; 2. Genomewide analysis: A. Design, B. Execution, C. Review and Critique; 3. Genetic molecular assays: A. Execution B. Interpretation; 4. Manuscript preparation: A. Writing of the first draft, B. Review and Critique. DZL: 1ABC, 2AC, 4A; DD: 2AB, 4B; NGM: 2AB, 4B; FT: 3AB, 4B; AA: 1BC, 4B; ES: 1ABC, 2AC, 4A.
Financial support
This study was supported by the National Institutes of Child Health and Human Development Grant, US, No HD 36071, to Dr DZ Loesch and Dr F Tassone, and by National Health and Medical Research Australia project grant No CF06/0269 to Prof E Storey, Dr DZ Loesch and Dr F Tassone.
Conflict of interest
None.
Ethical standards
This study was approved by the La Trobe University Human Research Ethics Committee (HEC01-85). All patients provided written informed consent, according to approved procedures. We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this work is consistent with those guidelines.






