



Introduction
Alzheimer's disease (AD) and Lewy body disease are the two commonest neurodegenerative causes of cognitive impairment. These often overlap, with some degree of AD co-pathology being present in many cases of Lewy body disease at autopsy (McAleese et al., Reference McAleese, Colloby, Thomas, Al-Sarraj, Ansorge, Neal and Attems2021). Antemortem, cases of dementia with Lewy bodies (DLB) are frequently positive for amyloid-β (Aβ) biomarkers, though less frequently tau positive (Ferreira et al., Reference Ferreira, Przybelski, Lesnick, Lemstra, Londos, Blanc and Kantarci2020).
Misdiagnosis is a particular concern early in the disease course, such as the prodromal ‘mild cognitive impairment’ (MCI) stage, when the full clinical picture may not yet be evident, and there is a greater likelihood of non-degenerative conditions being mistaken for early degenerative disease (e.g. depression-related cognitive dysfunctions, delirium, functional cognitive disorders, or misdiagnosed normal cognitive ageing). Recently developed plasma markers including AD specific and disease general biomarkers of astrocytosis and neuronal injury may help to address these diagnostic shortcomings (Zetterberg, Reference Zetterberg2019).
AD-specific markers may help to positively identify MCI due to AD (MCI-AD). There is a reduced ratio of amyloid-β (Aβ) 42 to Aβ40 in blood in both dementia due to AD and MCI-AD, and this reduction is associated with Aβ positivity on PET imaging (Nakamura et al., Reference Nakamura, Kaneko, Villemagne, Kato, Doecke, Doré and Yanagisawa2018). Concentrations of phosphorylated tau (p-tau) at threonine 181 (p-tau181), 217, or 231 are elevated in AD, including neuropathologically-confirmed cases and amyloid positive MCI-AD, relative to cognitively healthy individuals and many non-AD neurodegenerative diseases (Chong et al., Reference Chong, Ashton, Karikari, Tanaka, Schöll, Zetterberg and Lai2021). However, Aβ42/40 is also reduced, and p-tau181 elevated, in autopsy-confirmed DLB with AD co-pathology (Smirnov et al., Reference Smirnov, Ashton, Blennow, Zetterberg, Simrén, Lantero-Rodriguez and Galasko2022). MCI with Lewy bodies (MCI-LB) also features elevated p-tau181 relative to controls which may likewise reflect the presence of AD (Thomas et al., Reference Thomas, Hamilton, Heslegrave, Barker, Durcan, Lawley and O'Brien2022). However, not all cases of DLB feature AD co-pathology, though this is common (McAleese et al., Reference McAleese, Colloby, Thomas, Al-Sarraj, Ansorge, Neal and Attems2021). AD-specific biomarkers such as Aβ42/40 or p-tau may therefore be less useful in screening for Lewy body disease.
In contrast to the aetiology-specific AD biomarkers, plasma biomarkers of neurodegeneration which differentiate several neurological diseases from normal ageing may help to confirm the presence of progressive neurological disease in an ambiguous cognitive syndrome. Glial fibrillary acidic protein (GFAP), a protein expressed by astrocytes and possible marker of astrocytosis/astrocytic activation (Liddelow & Barres, Reference Liddelow and Barres2017), and neurofilament light chain (NfL), a marker of neuroaxonal injury (Karlsson, Rosengren, & Haglid, Reference Karlsson, Rosengren and Haglid1991), are elevated in several neurological diseases including, but not limited to AD and Lewy body disease (Gaetani et al., Reference Gaetani, Blennow, Calabresi, Di Filippo, Parnetti and Zetterberg2019; Schulz et al., Reference Schulz, Kruse, Gera, Kremer, Cedarbaum, Barbour and Mollenhauer2021). While potentially less specific to AD-related change than Aβ42/40 or p-tau, disease-general markers of astrocytosis/neurodegeneration may have greater utility at detecting other aetiologies in addition to AD (Chouliaras et al., Reference Chouliaras, Thomas, Malpetti, Donaghy, Kane, Mak and O'Brien2022).
We therefore aimed to assess whether MCI-LB featured a plasma biomarker profile of lowered Aβ42/40 ratio, increased phosphorylated tau, GFAP and NfL in comparison to healthy older adults, whether this differed from MCI-AD, and which markers best discriminated MCI and its subtypes from normal ageing. We hypothesised that:
(1) GFAP and NfL would best differentiate all-cause MCI, and MCI-LB alone, from normal ageing.
(2) Aβ42/40 ratio and p-tau181 would best differentiate MCI-AD from normal ageing.
(3) Aβ42/40 ratio and p-tau181 would be lowered and elevated respectively in MCI-AD relative to MCI-LB, but would not differentiate these reliably due to the variable and unknown prevalence of AD pathology in people with MCI-LB.
We also aimed to explore any longitudinal changes in plasma biomarkers over time, hypothesising that these may progressively worsen due to underlying disease progression.
Methods
Participants
Participants were drawn from two longitudinal cohorts as previously described (Donaghy et al., Reference Donaghy, Taylor, O'Brien, Barnett, Olsen, Colloby and Thomas2018, Reference Donaghy, Ciafone, Durcan, Hamilton, Barker, Lloyd and Thomas2022). Briefly, people aged ≥60 with a health service diagnosis of MCI were recruited from older person's memory, psychiatry, neurology and geriatric healthcare services in North East England. They were screened for inclusion, and retained if they did not have dementia, Parkinson's disease for >12 months prior to cognitive symptoms, a subjective-only cognitive impairment, or suspected frontotemporal or vascular aetiology.
Cognitively healthy comparators were recruited from friends and family of MCI participants, and through advertisement to local research involvement services.
All participants provided written, informed consent to participate.
Clinical assessment, imaging, and diagnosis
All participants underwent detailed clinical, cognitive and imaging assessment at baseline. Clinical and cognitive assessments were repeated at longitudinal follow-ups (annually prior to Covid-19, with an adaptive schedule thereafter). A three-person expert panel of old age psychiatrists (PCD, JPT, AJT) independently reviewed clinical research notes from each visit and provided a consensus diagnosis of MCI or all-cause dementia for each patient (Albert et al., Reference Albert, DeKosky, Dickson, Dubois, Feldman, Fox and Petersen2011; McKhann et al., Reference McKhann, Knopman, Chertkow, Hyman, Jack, Kawas and Mayeux2011). The same panel also independently rated the presence or absence of four core clinical features of Lewy body disease: complex visual hallucinations, fluctuating cognition, parkinsonian motor features, and REM sleep behaviour disorder (McKeith et al., Reference McKeith, Boeve, Dickson, Halliday, Taylor, Weintraub and Kosaka2017).
At baseline, all participants were offered 123I-N-fluoropropyl-2β-carbomethoxy-3β-(4-iodophenyl) single-photon emission computed tomography (FP-CIT) dopaminergic imaging (first cohort) or FP-CIT and 123I-metaiodobenzylguanidine (MIBG) cardiac scintigraphy (second cohort). FP-CIT images were visually rated blind to clinical information as normal or abnormal by a panel of experienced imaging analysts (Roberts et al., Reference Roberts, Donaghy, Lloyd, Durcan, Petrides, Colloby and Thomas2021a). Cardiac denervation was quantified as the ratio of MIBG uptake of the heart relative to the mediastinum (HMR) (Roberts et al., Reference Roberts, Durcan, Donaghy, Lawley, Ciafone, Hamilton and Thomas2021b) with an HMR abnormality cut-off < 1.85 based on assessment of our local healthy comparator sample (Kane et al., Reference Kane, Roberts, Petrides, Lloyd, O'Brien and Thomas2019).
Imaging and clinical characteristics were incorporated into clinical diagnosis to produce a differential classification of MCI as either MCI-AD (0 core clinical features of Lewy body disease, both biomarkers normal and fulfilling diagnostic criteria for MCI-AD) (Albert et al., Reference Albert, DeKosky, Dickson, Dubois, Feldman, Fox and Petersen2011), possible MCI-LB (1 core clinical feature of Lewy body disease with no abnormal biomarker, or 0 core clinical features of Lewy body disease but 1 + abnormal biomarker) or probable MCI-LB (2 + core clinical features of Lewy body disease, or 1 core clinical feature and 1 + abnormal biomarker) (McKeith et al., Reference McKeith, Ferman, Thomas, Blanc, Boeve, Fujishiro and Tiraboschi2020). These classifications were updated after each reassessment as new information came to light.
Biomarker assessment
Blood samples were collected at study baseline and at repeated follow-up by venepuncture in EDTA tubes, plasma was isolated by centrifuging, aliquoted and frozen at −80 °C for storage. Samples were collected either at the same visit as cognitive assessment, or at medical review which occurred a median of 13 days later.
Plasma samples were analysed by the UK Dementia Research Institute Biomarker Laboratory using Single molecule array (Simoa) HD1 analyser and the Quanterix Simoa Human Neurology 4-Plex E for Aβ40, Aβ42, GFAP and NfL. All samples were analysed with the same batch of reagents, at the same time. Plasma p-tau181 concentration was previously measured by this laboratory and reported in this cohort (Thomas et al., Reference Thomas, Hamilton, Heslegrave, Barker, Durcan, Lawley and O'Brien2022), and is included here for comparison.
The mean inter-assay % coefficients of variation (CV) were 3.80% for Aβ40, 3.17% for Aβ42, 6.13% for GFAP, and 4.24% for NfL.
Statistical analysis
Aβ40 and Aβ42 concentrations were combined to a ratio of Aβ42/40 as in previous research (Zetterberg, Reference Zetterberg2019). Anticipating that GFAP and NfL concentrations would follow a log-normal distribution as previously described (Chouliaras et al., Reference Chouliaras, Thomas, Malpetti, Donaghy, Kane, Mak and O'Brien2022), GFAP and NfL log concentrations were instead analysed. p-tau181 was likewise log transformed for analysis as previously described (Thomas et al., Reference Thomas, Hamilton, Heslegrave, Barker, Durcan, Lawley and O'Brien2022).
Diagnostic group differences (controls v. probable MCI-LB, and MCI-AD v. probable MCI-LB) were examined with linear models as previously (Thomas et al., Reference Thomas, Hamilton, Heslegrave, Barker, Durcan, Lawley and O'Brien2022), as were correlations between plasma markers and disease severity as assessed by the Addenbrooke's Cognitive Examination – Revised (ACE-R). In both cases, models were adjusted for age at baseline assessment. Longitudinal changes in biomarkers were assessed with linear or log-linear mixed models, including subject-level random intercept and correlated time slope, also adjusting for age at baseline. Correlations between plasma biomarkers were assessed with Pearson's correlation. Diagnostic classification analyses were undertaken using generalised linear models and the pROC package, with confidence intervals computed by DeLong's method.
Since possible MCI-LB is diagnostically uncertain, group comparisons did not include possible MCI-LB though raw data are included for these for further context, and they are included in all-cause MCI analyses.
All analyses were undertaken with R statistical software version 4.1.3. As an exploratory analysis, we did not adjust for multiple comparisons. Residuals of linear models were checked for normality with Q-Q plots.
Ethics and data sharing
This study received favourable ethical approval from the NRES Committee North East – Newcastle and North Tyneside 2 (12/NE/0290 and 15/NE/0420). The authors assert that all procedures contributing to this work comply with the ethical standards of the relevant national and institutional committees on human experimentation and with the Helsinki Declaration of 1975, as revised in 2008.
Data from these studies are available upon request through Dementias platform UK.
Results
Baseline characteristics of healthy controls and MCI sub-groups are presented in Table 1.
Table 1. Demographics and biomarker values at baseline for cognitively healthy older adults and MCI groups: Mean (s.d.) for continuous and Count (%) for categorical variables

Plasma markers
Group differences in plasma biomarkers and summary statistics are shown in Table 1 and Fig. 1.

Figure 1. Baseline plasma markers of Alzheimer's disease (Aβ42/40 ratio and p-tau181) and neurodegeneration (GFAP and NfL) in each diagnostic group: quartile boxplot plus 1.5 × IQR whiskers.
Plasma Aβ42/40 ratio did not significantly differ between probable MCI-LB and MCI-AD (Estimate = 0.001, [95% CI −0.003 to 0.005], p = 0.617) nor MCI-LB and healthy older adults (Estimate = 0.005 [−0.0002 to 0.0104], p = 0.063). MCI-AD did not have significantly lower Aβ42/40 ratio than controls (Estimate = −0.004 [−0.01 to 0.002], p = 0.170).
As previously reported, plasma p-tau181 was significantly elevated in probable MCI-LB relative to healthy older adults (Estimate = 0.31 [0.09 to 0.54], p = 0.007). p-tau181 was significantly higher in MCI-AD than both controls (Estimate = 0.52 [0.28 to 0.76], p < 0.001) and MCI-LB (Estimate = 0.21 [0.02 to 0.40], p = 0.033).
GFAP log concentration was significantly higher in probable MCI-LB cases than in healthy older adults (Estimate = 0.47 [0.22 to 0.71], p < 0.001). MCI-AD had significantly higher GFAP measures than both controls (Estimate = 0.71 [0.45 to 0.97], p < 0.001) and MCI-LB (Estimate = 0.24 [0.03 to 0.45], p = 0.028).
Log concentration of NfL in plasma was also significantly increased in probable MCI-LB relative to controls (Estimate = 0.25 [0.06 to 0.45], p = 0.012). MCI-AD also had significantly higher NfL than controls (Estimate = 0.36 [0.15 to 0.57], p = 0.001), but did not significantly differ from MCI-LB (Estimate = 0.11 [−0.06 to 0.28], p = 0.217).
We explored the possibility of differing ratios of ptau-181:NfL and ptau-181:GFAP, in both cases finding no significant differences between MCI-LB and MCI-AD (p = 0.382 and p = 0.522, respectively).
Pairwise associations between biomarkers were also assessed (see Fig. 2). Aβ42/40 was weakly negatively correlated with both plasma p-tau181 (r = −0.28, p < 0.001) and GFAP (r = −0.27, p = 0.001), but not significantly correlated with NfL (r = −0.05, p = 0.522). p-tau181 log concentration correlated strongly with GFAP log concentration (r = 0.63, p < 0.001), and moderately with NfL log concentration (r = 0.54, p < 0.001). Finally, GFAP and NfL log concentrations held a strong positive association (r = 0.67, p < 0.001).

Figure 2. Bivariate correlations between plasma markers of Alzheimer's disease (Aβ42/40 ratio and p-tau181) and neurodegeneration (GFAP and NfL).
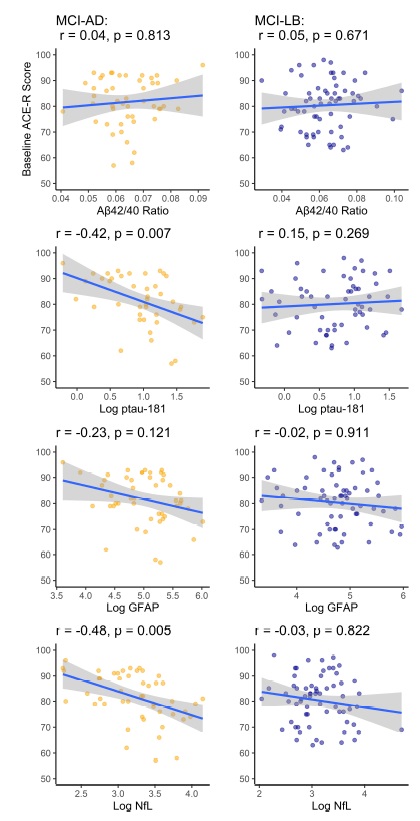
Association with baseline disease severity
Correlations between each plasma biomarker and baseline disease severity (total Addenbrooke's Cognitive Examination – Revised score; ACE-R) were examined with age-adjusted models within each MCI subgroup and overall.
In overall MCI, Aβ42/40 (r = 0.03, p = 0.755), GFAP (−0.12, p = 0.196), and p-tau181 (r = −0.07, p = 0.459) were not significantly associated with baseline age-adjusted ACE-R scores, while NfL held a weak negative association with baseline cognitive function (r = −0.24, p = 0.017).
Within MCI-AD Aβ42/40 (r = 0.04, p = 0.813) and GFAP (r = −0.23, p = 0.121) were not significantly associated with baseline ACE-R, but p-tau181 (r = −0.42 p = 0.007), and NfL (r = −0.48, p = 0.005) log concentrations held moderately significant correlations with greater disease severity.
Within probable MCI-LB, none of the plasma markers were significantly associated with baseline disease severity (see online Supplementary Fig. S1).
Discriminative utility
In discriminating any MCI case from healthy older adults, GFAP had the highest area under the ROC curve (AUC) with 0.75 [0.65 to 0.84], followed by p-tau181 (0.73 [0.63 to 0.84]), NfL (0.68[0.58 to 0.79]), and Aβ42/40 had the poorest discriminative utility (AUC of 0.64 [0.54 to 0.75]). GFAP was slightly superior to p-tau181 when sensitivity was favoured, and p-tau181 slightly superior when specificity was favoured (see Fig. 3).

Figure 3. Discriminative utility of each plasma biomarker at differentiating MCI from healthy older adults, MCI-AD from healthy, MCI-LB from healthy, and MCI-AD from MCI-LB.
Most biomarkers performed slightly better in discriminating MCI-AD from healthy controls (Aβ42/40 AUC = 0.63 [0.50 to 0.76], p-tau181 = 0.79 [0.68 to 0.91], GFAP = 0.82 [0.72 to 0.91], NfL = 0.72 [0.61 to 0.84]) and slightly worse in discriminating MCI-LB from controls (Aβ42/40 AUC = 0.63 [0.52 to 0.75], p-tau181 = 0.69 [0.57 to 0.82], GFAP = 0.71 [0.60 to 0.82], NfL = 0.67 [0.55 to 0.78]).
All plasma markers were generally poor at distinguishing MCI-AD from MCI-LB (Aβ42/40 AUC = 0.53 [0.42 to 0.64], p-tau181 = 0.62 [0.51 to 0.73], GFAP = 0.62 [0.51 to 0.72], NfL = 0.58 [0.47 to 0.69]).
Longitudinal change in biomarkers
Fifty-five MCI participants had provided two or more plasma samples to permit an analysis of any change over time. Mean (s.d.) time between first and final sample collection were 1.4 years (0.50) for MCI-AD, 1.4 years (0.57) for possible MCI-LB and 1.7 years (0.61) for probable MCI-LB.
In overall MCI, there was evidence of a marginally significant age-adjusted increase over time in Aβ42/40 (Estimate = +0.002 per year, p = 0.039) and NfL log-concentration (Estimate = +0.07 per year, p = 0.008) but not GFAP (Estimate = +0.01 per year, p = 0.675), or p-tau181 (Estimate = +0.04 per year, p = 0.305) (see Fig. 4).

Figure 4. Significant increases in Aβ42/40 ratio, and NfL concentration over time in MCI.
Discussion
We aimed to examine whether plasma biomarkers of AD (Aβ42/40 and p-tau181), astrocyte-expressed proteins (GFAP) and neurodegeneration (NfL) would differentiate MCI-LB from healthy controls, and MCI-AD from MCI-LB. We found evidence that both GFAP and NfL were elevated in MCI-LB, along with p-tau181 as previously described (Thomas et al., Reference Thomas, Hamilton, Heslegrave, Barker, Durcan, Lawley and O'Brien2022). Aβ42/40 was not clearly decreased in MCI-LB or MCI-AD.
Of the assessed markers, plasma GFAP best differentiated all-cause MCI from healthy controls, followed by p-tau181, NfL, with Aβ42/40 having the poorest utility. This same pattern was replicated within disease-specific subgroups, including MCI-AD. All markers were poor at differentiating MCI-LB from MCI-AD.
We also aimed to examine whether there were any changes in biomarker levels over time. We found a slight normalisation of Aβ42/40 ratio over time in MCI cases, and a slight increase in NfL over time.
These results suggest that in both MCI overall, and in disease-specific subgroups MCI-AD and MCI-LB, GFAP and p-tau181 differentiate between MCI cases and non-cases with similar accuracy. While AD-specific biomarkers correlate with amyloid and tau deposition (Chong et al., Reference Chong, Ashton, Karikari, Tanaka, Schöll, Zetterberg and Lai2021), these may not necessarily reflect consequential pathology in all cases and so may be relatively elevated in individuals with unrecognised asymptomatic AD within the healthy comparator group, reducing the specificity in differentiating MCI from healthy controls. Conversely, individuals without AD pathology may experience cognitive symptoms due to other processes (e.g. Lewy body disease or cerebrovascular disease), reducing the sensitivity to identify MCI.
GFAP, a disease-general marker of neurodegeneration, effectively differentiated both MCI-LB and MCI-AD from controls. This is consistent with previous research at the dementia stage which demonstrated that GFAP is elevated in both AD and Lewy body disease, though not in progressive supranuclear palsy or frontotemporal dementias (Chouliaras et al., Reference Chouliaras, Thomas, Malpetti, Donaghy, Kane, Mak and O'Brien2022), with the exception of progranulin-associated cases (Heller et al., Reference Heller, Foiani, Moore, Convery, Bocchetta and Neason2020); the latter aetiologies were excluded from these cohorts, and so these results may not translate to all MCI subtypes beyond MCI-AD and MCI-LB.
While NfL was less accurate than GFAP in distinguishing MCI from controls, this was moderately associated with age-adjusted disease severity in MCI. There was also evidence that levels of NfL in plasma were increasing over the course of MCI, suggesting that this may become more useful later in the disease course. GFAP, by contrast, remained stable. This may be consistent with the greater utility of NfL in distinguishing Lewy body disease from controls at the dementia stage (Chouliaras et al., Reference Chouliaras, Thomas, Malpetti, Donaghy, Kane, Mak and O'Brien2022), though this requires further exploration and validation.
GFAP and NfL were strongly correlated, consistent with previous research findings (Elahi et al., Reference Elahi, Casaletto, La Joie, Walters, Harvey, Wolf and Kramer2020), though possibly asynchronous, with NfL still increasing at this stage: previous research has also supported that NfL, but not GFAP, progressively increases over the course of MCI and dementia due to AD (Simrén et al., Reference Simrén, Leuzy, Karikari, Hye, Benedet, Lantero-Rodriguez and Ashton2021). Both measures were associated with p-tau181, but weakly correlated at best with Aβ42/40, suggesting that the former may have a closer association with astrocytic and neuronal dysfunction than the latter in MCI. These correlations are consistent with those seen in previous MCI and AD cohorts (Palmqvist et al., Reference Palmqvist, Stomrud, Cullen, Janelidze, Manuilova, Jethwa and Hansson2023).
There was an unexpected increase (i.e. normalisation) of Aβ42/40 over time in MCI, which is inconsistent with the expected worsening of underlying Aβ pathology. While previous data have been ambiguous as to whether Aβ42/40 ratio lowers over time in MCI (Simrén et al., Reference Simrén, Leuzy, Karikari, Hye, Benedet, Lantero-Rodriguez and Ashton2021), given the small effect size and marginal significance of this result we are not confident that this represents a genuine normalisation of Aβ42/40 over the course of MCI. This may instead reflect a plateauing effect of the underlying pathology at the prodromal/clinical transition.
None of the plasma biomarkers appeared to be useful in distinguishing MCI-AD from MCI-LB. This is consistent with the recognised prevalence of AD co-pathology in DLB at autopsy (McAleese et al., Reference McAleese, Colloby, Thomas, Al-Sarraj, Ansorge, Neal and Attems2021), and supports that this may also be present early in MCI-LB. Whether this co-pathology is sufficient to affect the clinical manifestation or disease progression at this stage is less clear: we have previously demonstrated that p-tau181 concentration is associated with disease progression in MCI-AD, but not MCI-LB (Thomas et al., Reference Thomas, Hamilton, Heslegrave, Barker, Durcan, Lawley and O'Brien2022). In contrast, when dementia is clinically manifest, lower Aβ42/40 is associated with disease progression in DLB (Chouliaras et al., Reference Chouliaras, Thomas, Malpetti, Donaghy, Kane, Mak and O'Brien2022).
These findings highlight the need for sensitive markers of alpha-synuclein pathology; while available biomarkers such as FP-CIT and MIBG are highly specific to DLB and MCI-LB, the sensitivity is lower than at the dementia stage (Roberts et al., Reference Roberts, Donaghy, Lloyd, Durcan, Petrides, Colloby and Thomas2021a, Reference Roberts, Durcan, Donaghy, Lawley, Ciafone, Hamilton and Thomas2021b). We also demonstrate here that AD and neurodegeneration plasma biomarkers do not reliably differentiate MCI-AD from MCI-LB. Synuclein assays have been recently shown to detect prodromal and clinically manifest Lewy body disease in cerebrospinal fluid (Iranzo et al., Reference Iranzo, Fairfoul, Ayudhaya, Serradell, Gelpi, Vilaseca and Green2021), skin punch samples (Mammana et al., Reference Mammana, Baiardi, Quadalti, Rossi, Donadio, Capellari and Parchi2021), and recently in blood (Kluge et al., Reference Kluge, Bunk, Schaeffer, Drobny, Xiang, Knacke and Zunke2022): disease-specific synuclein assays may therefore offer a method of reliably differentiating MCI-LB from MCI-AD, when more clinically accessible (i.e. blood-based) biomarkers are available.
Strengths and limitations
This study benefitted from the inclusion of a clinically well-characterised cohort, with several years of follow-up, and differentially classified as MCI-AD or MCI-LB using current consensus criteria. However, as participants were recruited at the early stages of disease, autopsy validation of clinical diagnoses are not available for the majority of individuals, though follow-up and approach for brain tissue donation are ongoing.
Similarly, we did not have confirmatory CSF or PET testing available for Aβ or tau. Previous work has typically examined the utility of these biomarkers to differentiate Aβ-positive cases from Aβ-negative cases of MCI or dementia, and so we are unable to directly compare our AUC values to these (Benedet et al., Reference Benedet, Milà-Alomà, Vrillon, Ashton, Pascoal, Lussier and Suárez-Calvet2021; Chouliaras et al., Reference Chouliaras, Thomas, Malpetti, Donaghy, Kane, Mak and O'Brien2022; Palmqvist et al., Reference Palmqvist, Stomrud, Cullen, Janelidze, Manuilova, Jethwa and Hansson2023).
We present confidence intervals and p values unadjusted for multiple comparisons, and so encourage caution in interpreting these. While several reported estimates would be robust to any such adjustment, other estimates have a high degree of uncertainty: confidence intervals around the plasma biomarker AUCs are wide, and so these require further replication. Fewer participants had longitudinal biomarker measurements available, limiting the capability to assess disease-specific trajectories meaningfully.
While we have previously shown both MCI groups in these cohorts to reliably decline in cognition (Hamilton et al., Reference Hamilton, Matthews, Donaghy, Taylor, O'Brien, Barnett and Thomas2021b, Reference Hamilton, Matthews, Donaghy, Taylor, O'Brien, Barnett and Thomas2021c), and convert to dementia (Hamilton et al., Reference Hamilton, Matthews, Donaghy, Taylor, O'Brien, Barnett and Thomas2021a), it is possible that an unknown number of these MCI cases may have a non-degenerative aetiology, such as a functional cognitive disorder mimicking MCI-AD or MCI-LB (Ball et al., Reference Ball, McWhirter, Ballard, Bhome, Blackburn, Edwards and Carson2020). Such unrecognised cases might not be expected to feature either an AD-like or broadly neurodegenerative biomarker profile, and so may lower the accuracy of these plasma biomarkers in this setting. Future research may benefit from assessing the relationship between these biomarkers and the longitudinal course of disease progression to assess whether the use of these might improve the identification of a progressive neurodegenerative MCI.
Conclusions
GFAP assays performed comparably to p-tau181 in differentiating MCI from healthy controls, both overall and within MCI-AD or MCI-LB specifically. Markers of neurodegeneration or astrocytic protein expression may therefore help identify symptomatic neuropathological changes in AD and other diseases. Plasma AD and neurodegenerative markers had limited utility in differentiating MCI-AD from MCI-LB, highlighting a need for synucleinopathy-specific biomarkers.
Supplementary material
The supplementary material for this article can be found at https://doi.org/10.1017/S0033291723001952.
Acknowledgements
None.
Financial support
This work was supported by Alzheimer's Research UK (ARUK-PG2015-13) and by the NIHR Newcastle Biomedical Research Centre. GE Healthcare provided the FP-CIT ligand for this investigator-led study. H. Z. is a Wallenberg Scholar supported by grants from the Swedish Research Council (#2018-02532), the European Union's Horizon Europe research and innovation programme under grant agreement No 101053962, Swedish State Support for Clinical Research (#ALFGBG-71320), the Alzheimer Drug Discovery Foundation (ADDF), USA (#201809-2016862), the AD Strategic Fund and the Alzheimer's Association (#ADSF-21-831376-C, #ADSF-21-831381-C, and #ADSF-21-831377-C), the Bluefield Project, the Olav Thon Foundation, the Erling-Persson Family Foundation, Stiftelsen för Gamla Tjänarinnor, Hjärnfonden, Sweden (#FO2022-0270), the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No 860197 (MIRIADE), the European Union Joint Programme – Neurodegenerative Disease Research (JPND2021-00694), and the UK Dementia Research Institute at UCL (UKDRI-1003). P. C. D. is supported by the Medical Research Council (grant number MR/W000229/1).
Competing interest
C. A. H. has received honoraria from Dementias platform UK for educational activities unrelated to this work. J. T. O'B has acted as a consultant for TauRx, Novo Nordisk, Roche and GE Healthcare and received grant support from Merck and Alliance Medical, all unrelated to this work. G. R. has received honoraria from GE Healthcare for educational presentations. J.-P. T. has acted as consultant for Sosei Heptares and Kyowa-Kirin, and has received speaker fees from GE Healthcare and Bial, all unrelated to this work. H. Z. has served at scientific advisory boards and/or as a consultant for Abbvie, Acumen, Alector, Alzinova, ALZPath, Annexon, Apellis, Artery Therapeutics, AZTherapies, CogRx, Denali, Eisai, Nervgen, Novo Nordisk, Passage Bio, Pinteon Therapeutics, Prothena, Red Abbey Labs, reMYND, Roche, Samumed, Siemens Healthineers, Triplet Therapeutics, and Wave, has given lectures in symposia sponsored by Cellectricon, Fujirebio, Alzecure, Biogen, and Roche, and is a co-founder of Brain Biomarker Solutions in Gothenburg AB (BBS), which is a part of the GU Ventures Incubator Program (outside submitted work). All other authors report no conflict of interests.








 Open access
Open access