


Fingolimod (Gilenya®) is the first oral therapy to be approved in Canada and is widely used for the treatment of relapsing-remitting multiple sclerosis (RRMS) to reduce the frequency of clinical exacerbations and to delay the progression of physical disability. Clinical efficacy in RRMS was demonstrated in two phase III placebo-controlled trials (FREEDOMS, FREEDOMS II), and a phase III comparator trial versus intramuscular interferonβ-1a (TRANSFORMS).Reference Kappos, Radue and O’Connor 1 - Reference Cohen, Barkhof and Comi 3
With respect to safety, the incidence of severe bradycardia in the FREEDOMS trial was 0.9% with fingolimod compared to 0.2% with placebo.Reference Kappos, Radue and O’Connor 1 Accordingly, a six-hour observation period is required to identify patients who are more susceptible to cardiac effects during treatment initiation. 4 The incidence of macular edema was 0.5%, with most cases occurring in the first four months after fingolimod exposure,Reference Kappos, Radue and O’Connor 1 and an ophthalmic examination is recommended at three to four months after treatment initiation. 4
The GILENYA® Go ProgramTM was launched in Canada in March 2011 to provide patient support services, reinforce patient adherence to recommended monitoring and coordinate first-dose observation (FDO) at dedicated FDO centers. By virtue of the Go ProgramTM structure, nearly all Canadian patients prescribed fingolimod are captured in the database. Few patients may not be registered due to a lack of physician and/or patient awareness of the program or requirement for assistance. The program comprises approximately 100 FDO sites and 194 ophthalmic clinics across Canada.
Candidates for fingolimod are enrolled in the patient support program by their treating neurologist after providing informed consent and receive a follow-up call from a nurse navigator specialized in multiple sclerosis (MS) within 24 hours. A reimbursement specialist investigates drug reimbursement (public and/or private insurance). A nurse navigator collaborates with the treating neurologist, MS center staff and the patient to coordinate the administration of the first dose at an MS clinic or FDO center. Patients receive a telephone reminder 24 hours prior to all scheduled FDO, ophthalmic exam and other appointments. There is also a patient web portal available for self-reminders. Following initiation of fingolimod at an FDO center, the treating neurologist receives an FDO report that summarizes any clinical findings, adverse events, and electrocardiogram (ECG) results.
The support program also coordinates medication reimbursement, pharmacy services and dispensing of fingolimod by a local retail or Go ProgramTM network pharmacy. The nurse navigator schedules the follow-up ophthalmic examination and reminds the patient of the appointment. The results of the ophthalmic examination are forwarded from the ophthalmic clinic to the neurologist.
Data are collected from patient contacts with program staff and the attending healthcare providers and entered into the Go ProgramTM database. The present analysis evaluated patient adherence to treatment and monitoring after fingolimod initiation, drug tolerability and adverse effects of interest during the first three years of the program. Adverse events were those reported by health care providers or patients and other adverse events may have occurred.
Method
Data were collected and analyzed for patients enrolled in the Canadian GILENYA® Go ProgramTM from the time of first patient enrollment in March 2011 to March 31, 2014. Subjects were excluded if they withdrew from the program prior to receiving the first dose or had not yet taken their first dose at the time of data cut-off. Reports of adverse effects were validated with a database of adverse events reported to Novartis Pharmaceuticals Canada Inc.
Results
Demographics
A total of 2,399 patients had completed FDO at the end of the three-year observation period (Table 1). Mean age was 41.2 years, and 75.2% were female. The majority of patients resided in the provinces of Quebec (49.1%), Ontario (23.3%), Alberta (13.8%) and British Columbia (6.9%), with the remainder in Nova Scotia, New Brunswick, Manitoba, Newfoundland and Labrador, Saskatchewan and Prince Edward Island.
Table 1 Demographics of patients enrolled in the GILENYA® Go ProgramTM.

In Canada, fingolimod is generally prescribed to RRMS patients who have experienced a suboptimal response or poor tolerability with a previous disease-modifying therapy (DMT). 4 The most recent prior therapies reported by patients (n=2271) were glatiramer acetate (31.1%), subcutaneous interferon β-1a (25.2%), intramuscular interferon β-1a (14.9%), natalizumab (14.2%), subcutaneous interferon β-1b (10.1%), and other (1.1%) (Figure 1). Prior use of dimethyl fumarate or teriflunomide was low due to the recent approval of these agents in Canada. Fingolimod was the first-line treatment in 76 patients (3.3%). Patient-reported reasons for switching to fingolimod (n=1848) were lack of efficacy (34.9%), side effects (34.6%), and dissatisfaction with injections/infusion (30.5%) (Figure 2).
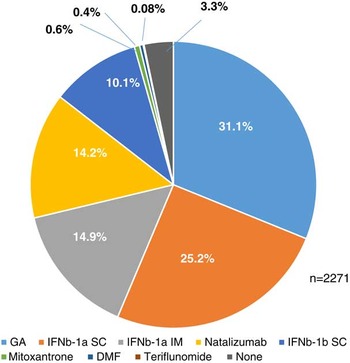
Figure 1 Most recent prior therapies reported by patients in the Go program database.

Figure 2 Reason for switching from prior therapy to fingolimod by patient self-report.
Continuation rate
At data cut-off, 2,072 patients were currently in treatment and 327 had withdrawn, for an overall continuation rate of 86.4% (Figure 3). The 12-month continuation rate (n=1392) was 80.7%. The 24-month continuation rate (n=778) was 76.6%, and the 30-month continuation rate (n=350) was 76.0%. The proportion of patients successfully maintained on fingolimod was similar for those previously treated with intramuscular interferon β-1a (88.2%), subcutaneous interferon β-1a (86.5%), glatiramer acetate (86.3%), natalizumab (85.1%), and subcutaneous interferon β-1b (84.3%) but was lower for those most recently treated with mitoxantrone (69.2%).

Figure 3 Cumulative continuation rate in the first 36 months of the GILENYA® Go ProgramTM.
A subset of 93 patients (3.9%) underwent a second FDO during the observation period. The most common reasons for treatment interruption were patient or physician request (n=14), abnormal blood test results (n=12), gastrointestinal symptoms (n=8), desire to become pregnant (n=8), and headaches (n=5). Following the second FDO, 80 of 93 patients (86.0%) were successfully maintained on fingolimod, with a mean treatment duration of 298 days at data cut-off.
Discontinuations and adverse effects
Among the 327 patients (13.6%) in the database who discontinued therapy, the average time to withdrawal was 181 days. Females (mean age 42.7 years) were more likely than males (mean age 42.8 years) to withdraw from therapy (14.6% vs. 10.7%). Treatment withdrawal rates were highest in Ontario (15.8%), Quebec (15.5%) and British Columbia (12.1%), and <10% in all other provinces. Withdrawals were generally more common with increasing age. The withdrawal incidence was 5.0% for those aged 18-20 years; 11.7% for those aged 20 to <30 years; 11.8% for those aged 30 to <40 years; 14.0% for those age 40 to <50 years; 18.4% for those aged 50 to <60 years; and 11.4% for those aged ≥60 years.
The most common reasons for discontinuation (n=323) were adverse effects (59.8%), physician request (13.3%), lack of efficacy (9.3%), and patient request (8.7%) (Figure 4). It should be noted that during the observation period, there was a revision to the fingolimod product label to provide additional guidance concerning patient selection for fingolimod in Canada. A total of 120 of 327 withdrawals (36.7%) occurred during this period (February 2012 – September 2012). This represented 45.1% of all withdrawals due to physician or patient request, which may or may not have been related to the safety, tolerability or effectiveness of fingolimod.

Figure 4 Reasons for fingolimod discontinuation.
For the full cohort, the most common adverse effects associated with discontinuation were low lymphocyte count/abnormal blood work (7.0%), elevated liver enzymes (4.9%), gastrointestinal disturbance (3.4%), headache/migraine (3.4%), and palpitations (2.4%). Most discontinuations occurred in days 30-179 of treatment (Figure 5).
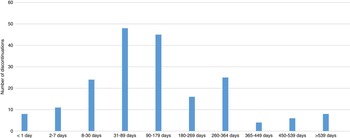
Figure 5 Time course of adverse events leading to discontinuation.
Serious adverse events included breast cancer (n=2, days 288 and 692), Bowen’s disease (n=1, day 453), cervical cancer (n=1, day 184), kidney cancer (n=1; day 14), pericardial effusion (n=2, day 325), left ventricular dysfunction (n=1; day 670), arrhythmia (n=1; day 165), and pulmonary emboli (n=1; day 304). The overall incidence of cancers was 0.21%. There was one death in a 55-year-old female on day 484 of treatment. The cause of death has not been confirmed.
Other adverse events of interest associated with treatment discontinuation during the three-year reporting period included lower respiratory tract infection (n=10), difficulty breathing (n=9), chest pain (n=8), hypertension (n=7), MS relapse (n=6), and herpes zoster infection (n=5). The overall rate of herpesvirus infections was 1.9%. No cases of progressive multifocal leukoencephalopathy (PML) were reported.
Two cases of macular edema (on days 8 and 22) were not attributed to treatment. During the course of therapy, there were 11 additional cases of macular edema (0.46%), and two cases of uveitis (0.08%). Ophthalmic examinations were reported for 12 of 13 cases. Treatment was continued in three and discontinued in nine patients; data were missing for one case. In one case, macular edema was diagnosed prior to the scheduled ophthalmic examination and treatment was discontinued. In five cases, abnormalities were detected at the ophthalmic examination and treatment was subsequently withdrawn. In three cases, the examination was performed in week 15 of treatment and abnormalities were reported >25 weeks afterward, at which time treatment was withdrawn. A delay in reporting of macular edema may be explained by a delay in confirmation of symptoms by an ophthalmologist. For the eight cases of macular edema leading to discontinuation for which data were available, the average time from FDO to stopping therapy was 209 days (range 50-460 days).
There were 19 early withdrawals (0.79%), defined as less than seven days after FDO. Reasons for withdrawal at first dose (days one to two, n=12) were first-degree atrioventricular (AV) block (n=4), second-degree AV block (n=3), AV block unspecified (n=1), bradycardia (n=2), migraine (n=1) and itchiness (n=1). For the full dataset, the overall incidence of AV block (second-degree or higher) was 0.17%. The incidence of all cardiac-related events (AV block, bradycardia, chest pain, dizziness) leading to early withdrawal was 0.5%.
Discontinuation due to lack of efficacy
A total of 30 patients (1.3%) starting treatment with fingolimod discontinued due to self-reported lack of efficacy. The mean time to discontinuation due to lack of efficacy was 296.5 days (range 14-753 days). Six of these 30 patients had discontinued their previous treatment due to lack of efficacy while on subcutaneous interferon β-1a, glatiramer acetate or natalizumab. Eight patients had received prior treatment with natalizumab or mitoxantrone. Three patients were previously untreated.
Patient support program effectiveness
Following enrollment in the GILENYA® Go ProgramTM, a nurse navigator contacts the patient and schedules the FDO appointment and any pre-FDO assessments, as required. A follow-up ophthalmic examination is recommended at three to four months post-initiation. Data were available for 1,998 patients on treatment and scheduled for an ophthalmic examination (minimum 120 days on treatment). Overall, there were 1,887 reports (94.4%) of patients attending the scheduled ophthalmic examination, and 1,761 (88.1%) had the examination ≤120 days post FDO. The mean time from FDO to ophthalmic examination was 102.8 days. Ophthalmic examinations not performed through the GILENYA® Go ProgramTM were not recorded in the database.
Conclusions
The GILENYA® Go ProgramTM demonstrates that a highly structured patient program providing education, assistance to secure reimbursement and appointment scheduling is an effective means of supporting adherence to therapy and monitoring requirements for RRMS patients. Adherence is also reinforced by ongoing daily reminders to take the medication sent to patients by email and/or SMS texting. Ongoing support by nurse navigators and other program personnel may have contributed to the high treatment continuation rate. Indeed, the two-year continuation rate of 76.6% was comparable to the 75.3% continuation rate seen in the two-year FREEDOMS trial.Reference Kappos, Radue and O’Connor 1 The perceived effectiveness and tolerability of fingolimod may have also contributed to the high continuation rate in the program, with a recent single-center analysis reporting a lower discontinuation rate at three months with fingolimod versus dimethyl fumarate (8% vs. 18%), which was largely attributed to a lower dropout rate due to poor tolerability with fingolimod.Reference Cohn, Bermel and Hara-Cleaver 5 The group subsequently reported a one-year continuation rate with fingolimod of 72.5%,Reference Hersh, Hara-Cleaver, Rudick, Cohen, Bermel and Ontaneda 6 which was lower than the 80.7% rate at one year reported in this analysis.
The Go ProgramTM continuation rate at three years is higher than the cumulative persistence rate at two years of 41.5-47.4% with injectable DMTs seen in real-world practice in Canada.Reference Wong, Gomes, Mamdani, Manno and O’Connor 7 A comparison with the natalizumab continuation rate cannot be made since patients frequently discontinue natalizumab for reasons other than poor tolerability, such as anti-JC virus antibody status, duration of treatment, and abnormal laboratory values. It was also noteworthy that the Go database represents generally older RRMS patients than those enrolled in controlled clinical trials and most had received a prior DMT, including 14.2% who had switched from natalizumab. Patients may have been more motivated to remain on treatment or to resume fingolimod after treatment interruption because of prior treatment failure. However, the database was not designed to capture the reasons for the rates of adherence and persistence.
First-dose administration was well tolerated, with a withdrawal rate at FDO of less than one percent. The overall incidence of second-degree or higher AV blocks with fingolimod 0.5 mg was 0.17%. This was somewhat lower than the 0.2% rate that has been previously reported in a pooled analysis of phase III trials.Reference DiMarco, O’Connor and Cohen 8
The 0.46% incidence of macular edema was similar to the 0.5% rate reported in the pooled safety analysis of phase II and III trials.Reference Jain and Bhatti 9 The mean time to reporting of macular edema was about seven months.
Adverse effects associated with treatment discontinuation were similar to what has been seen in the clinical trial setting and included elevated liver enzymes, gastrointestinal symptoms and headache. Low lymphocyte counts, which occur as part of the mechanism of action of fingolimod, were also a common reason for discontinuing therapy. Most withdrawals due to adverse effects occurred in the first six months of treatment, suggesting that more intensive efforts at managing adverse effects, such as gastrointestinal upset, headache and fatigue, in the initial months of therapy may help to promote adherence.
The database analysis has a number of limitations. Due to patient privacy, the GILENYA® Go ProgramTM has limited access to clinical information, such as comorbidities, concomitant therapies, duration of RRMS, duration of prior DMT use and JC virus antibody status, which may have provided information on the patients who initiated fingolimod and their individual need for support and education. Patients enrolled in the program represent those seen in routine clinical practice and this cohort may differ from those studied in clinical trials. Information reported to the program by health care providers and patients may be incomplete; with respect to adverse event reporting, events may be over- or underreported by patients, or may not have been medically confirmed. In addition, no follow-up information is obtained after a patient withdraws from the program, with the exception of the three to four month ophthalmic exam appointment.
While patient support databases have their limitations, obtaining information in this manner may provide important insights on the patient experience after initiating a treatment for RRMS. This analysis of the GILENYA® Go ProgramTM database indicates that patients can successfully complete the required six-hour observation period, and can be satisfactorily managed over the longer term, as evidenced by the high rate of continuation over the first three years of therapy.
Interim analyses were reported in
Lapierre Y, O’Connor P, Schecter R. Long-term treatment monitoring. Presented at the 29th Congress of the European Committee on Treatment and Research in Multiple Sclerosis (ECTRIMS), Copenhagen, Denmark, 2-5 October 2013; abstract P1033.
Lapierre Y, Selchen D, Devonshire V, Schecter R. Switching from injectable therapy leads to high continuation rate on fingolimod. Presented at the American Academy of Neurology annual meeting, 26 April-3 May 2014, Philadelpha PA, USA; abstract P7.221.
Freedman MS, Burton J, Schecter R. Real-life patient adherence and follow-up monitoring with fingolimod. Presented at the Canadian Neurological Sciences Federation 49th Congress, 3-6 June 2014, Banff, Alberta, Canada; abstract P033.
Freedman MS, O’Connor P, Duquette P, Schecter R. High continuation rate, good disease control after switching from natalizumab to fingolimod. Presented at the Consortium of Multiple Sclerosis Clinics (CMSC)-Americas Committee for Treatment and Research in Multiple Sclerosis (ACTRIMS) Cooperative Annual Meeting, Dallas, TX, USA, 28-31 May 2014; abstract DX48.
Acknowledgements
The authors take full responsibility for the content of the paper and thank Steven Manners of Communications Lansdowne for editorial assistance under their guidance. Editorial help was funded by Novartis Pharmaceuticals Canada Inc. We thank Catherine Dickinson, Kelly Anderson and Sonia Fendler from the GILENYA® Go ProgramTM for providing excellent support.
DISCLOSURES
Yves Lapierre does not have anything to disclose.
Paul O’Connor has the following disclosures:Biogen Idec, Consultant, Consulting fees and research support
Virginia Devonshire has the following disclosures:
Novartis, Consultant, Consulting fees
Mark S. Freedman has the following disclosures:
Biogen Idec, Consultant/Advisor, Consulting fees/Honoraria
Bayer Healthcare, Consultant/Advisor, Consulting fees/Honoraria
EMD Canada, Consultant/Advisor, Consulting fees/Honoraria
Genzyme, Consultant/Advisor, Consulting fees/Honoraria
Novartis, Consultant/Advisor, Consulting fees/Honoraria
Roche, Consultant/Advisor, Consulting fees/Honoraria
Teva Canada Innovation, Consultant/Advisor, Consulting fees/Honoraria
Chugai, Adjudication Committee, Honoraria
Genzyme, Speaker’s Bureau, Honoraria
Marcelo Kremenchutzky has the following disclosures:
Biogen Idec, Grant recipient and consultant, Grants and consulting fees
Bayer, Researcher, Research grants
Genzyme, Grant recipient and consultant, Research grants and consulting fees
Sanofi, Researcher, Research grant
Novartis, Grant recipient and consultant, Grants and consulting fees
Teva, Researcher, Research grants and consulting fees
Society of Canada, Operations, Operational grants
CIHR, Researcher, Research grant
Michael Yeung has the following disclosures:
Novartis, Consultant/Advisor, Consulting fees
Novartis, Researcher, Research support
Teva Canada Innovation, Consultant/Advisor, Consulting fees
Teva Canada Innovation, Researcher, Research support
EMD Serono, Consultant/Advisor, Consulting fees
Genzyme, Consultant/Advisor, Consulting fees
Genzyme, Researcher, Research support
Biogen Idec, Researcher, Research support
Hoffmann-La Roche, Researcher, Research support
CADTH/Health Canada, Consultant/Advisor, Consulting fees
Robyn Schecter has the following disclosures:
Novartis, Employee, Salary







 Open access
Open access