

Collagen, cellulose, chitin, and DNA are abundant natural polymers that self-assemble in aqueous environments into stiff and strong helices. Among those, DNA stands out due to its well-known double-strand structure, a feature that is challenging to obtain with synthetic polymers, which usually lack the specific interactions and conformations that are characteristic of nucleic acids and peptides. The availability of synthetic polymers that can self-assemble into double-stranded helices in solution would open the door to greater understanding of macromolecular self-assembly mechanisms, new applications, and provide a cheap, scalable, and sustainable source of reinforcing fibers for composite materials.
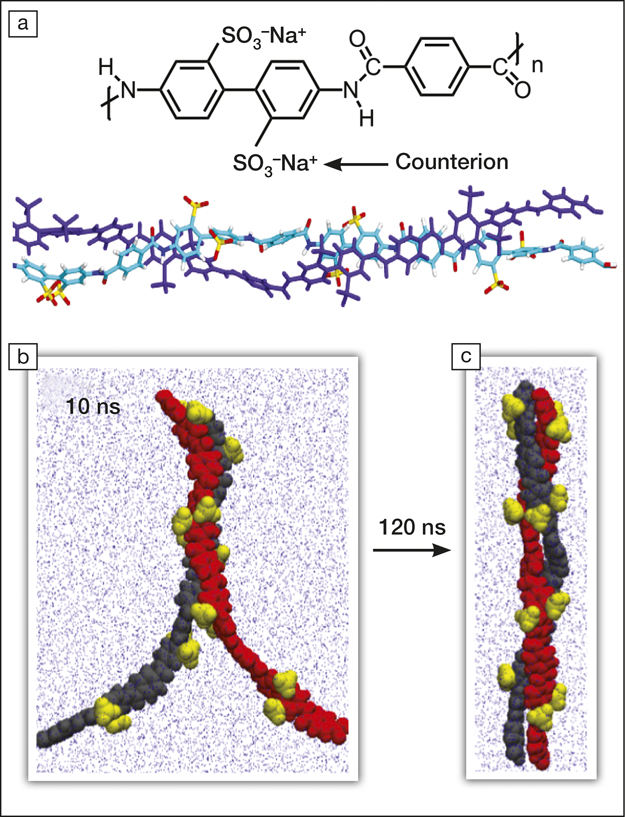
(a) The chemical unit of poly-2,2´-disulfonyl-4,4´-benzidine terephthalamide (PBDT) and the molecular dynamics simulation of two PBDT dimers. (b, c) Molecular dynamics snapshots (b) after 10 ns of 2 PBDT monomers self-assembling in water and (c) after 120 ns. The atoms from each monomer are represented separately as black and red; the yellow balls indicate the sulfonate (SO3–) groups at the surface of the polymer.
This is the challenge that attracted the research team led by Louis Madsen, Rui Qiao, and Theo Dingemans at Virginia Tech and The University of North Carolina at Chapel Hill, in the United States, and at Delft University of Technology and the University of Amsterdam, in The Netherlands. Using a polymer called poly-2,2´-disulfonyl-4,4´-benzidine ter-ephthalamide (PBDT), they were able to show that PBDT forms a double helix with a high stiffness. They reported their results in a recent issue of Nature Communications (doi:10.1038/s41467-019-08756-3).
Although it is possible to measure the Young’s modulus of DNA using nano-mechanical tests, one convenient way to describe the stiffness of a short organic molecule is by determining its persistence length. The persistence length describes the shortest length below which the polymer behaves like a rigid beam. Above this length, the polymer behaves like soft spaghetti. In the case of PBDT, the persistence length is of the order of 1 µm, which is nearly 20 times longer than for DNA, and hence 20 times stiffer.
In their study, the researchers used nuclear magnetic resonance (NMR) spectroscopy and x-ray diffraction to measure the persistence length and characterize the double helical structure. They suspended preformed PBDT double helices in water with Na+ counterions and observed their alignment into ordered nematic phases similar to liquid crystals in optical displays. Using a magnetic field of 500 mT, they oriented all of the micro-meter-sized nematic domains along the same direction to record the NMR spectrum and the diffraction pattern.
The researchers also used spectroscopic methods to investigate the dynamics and alignment of the helix, which supported their findings using molecular dynamics simulations. The self-assembly of PBDT, driven by enthalpic and entropic effects that result from electrostatic repulsions, hydrophobic stacking of phenyl groups, and interactions between amide groups, occurs within 30 ns (see Figure).
Gustav Nyström, head of the Laboratory for Cellulose & Wood Materials at Empa—Swiss Federal Laboratories for Materials Science and Technology, emphasizes that “the results are interesting since the PBDT double helix reaches unusually long persistence lengths approaching those of the 2 µm rigid segments of nanocellulose fibrils, and 1–3 µm regions of amyloid fibrils. This observation suggests that these helical assemblies may, in common with nanocellulose or amyloids, be interesting as building blocks for functional composite materials.” Nyström is also looking forward to a deeper look into the assembly mechanism and the interactions at play with PBDT.
Coincidently, these are two directions the research team led by Madsen, Qiao, and Dingemans are taking. In addition to investigating the molecular interactions, they are also “making controlled changes to the PBDT molecular structure and to the synthesis protocols to study how long the persistence length can be, what are the key interactions and how the helix originally forms,” says Madsen. “Our team is also pursuing many other avenues related to the PBDT rods, such as making ionic composite electrolytes with high stiffness and liquid-like ionic conductivity, and structural composites with high strength and small filler amounts.”
Indeed, one advantage of using synthetic chemistry over using biological fibers is the possibility of obtaining large quantities using inexpensive and scalable chemical reactions. Madsen further explains that “it is very easy to make 10–100 g quantities in the lab from inexpensive starting materials that are commercially available. We expect we can very easily scale this up to kg quantities.”
