



The rise of the α-helix
Linus Pauling in 1950 published a three-dimensional model for a universal protein secondary structure motif which he initially called the α-spiral (Pauling and Corey, Reference Pauling and Corey1950; Dunitz, Reference Dunitz2001). Jack Dunitz, then a postdoc in Pauling's lab suggested to Pauling that the term helix is more accurate than spiral when describing the right-handed peptide and protein coiled structures. Pauling agreed, hence the rise of the α-helix (Pauling et al., Reference Pauling, Corey and Branson1951; Dunitz, Reference Dunitz2013; Egli and Zhang, Reference Egli and Zhang2022), and, by extension, the ‘double helix’ structure of DNA. Although structural biologists and protein chemists are familiar with varying polar and apolar characters of amino acids in α-helices (Eisenberg et al., Reference Eisenberg, Weiss and Terwilliger1982; Eisenberg et al., Reference Eisenberg, Weiss and Terwilliger1984; Eisenberg, Reference Eisenberg2003), to non-experts the three chemically distinct α-helix types classified here may hide in plain sight.
Three helix variants in proteins
There are three variants of helices found in proteins (Stryer, Reference Stryer1981; Brändén and Tooze, Reference Brändén and Tooze1991; Fersht, Reference Fersht1998; Fodje and Al-Karadaghi, Reference Fodje and Al-Karadaghi2002; Armen et al., Reference Armen, Alonso and Daggett2003; Liljas et al., Reference Liljas, Liljas, Ash, Lindblom, Nissen and Kjeldgaard2017). Briefly, the most commonly observed α-helix has 3.6 residues per 360° helical turn (100° rotation per amino acid) and exhibits a translation of 1.5 Å along the helical axis with i, i + 4 hydrogen bond (H-bond) formation (Pauling and Corey, Reference Pauling and Corey1950; Pauling et al., Reference Pauling, Corey and Branson1951). There are two additional kinds of helices: The 310-helix has 3 residues per 360° helical turn (120° rotation per amino acid) and exhibits a translation of 2 Å along the helical axis with i, i + 3 H-bonding (Bragg et al., Reference Bragg, Kendrew and Perutz1950; Perutz et al., Reference Perutz, Rossmann, Cullis, Muirhead and Will1960). This 310-helix often occurs at the end of α-helices (Armen et al., Reference Armen, Alonso and Daggett2003). The π-helix has 4.4 residues per 360° helical turn (83° rotation per amino acid) and exhibits a translation of 1.2 Å along the helical axis with i, i + 5 H-bonding (Fodje and Al-Karadaghi, Reference Fodje and Al-Karadaghi2002). The π-helix occurs in the middle of longer α-helices (Armen et al., Reference Armen, Alonso and Daggett2003). However, our analysis is not about the variations in the geometry of protein helices; interested readers should instead consult the original and more recent literature. Rather, we propose to classify structurally identical α-helices into three chemically distinct types, namely: Type I, hydrophilic α-helix; Type II, hydrophobic α-helix; Type III, amphiphilic α-helix.
Twenty amino acids with hydrophilic and hydrophobic characteristics
There are two general classes of amino acids, hydrophilic and hydrophobic (Stryer, Reference Stryer1981; Brändén and Tooze, Reference Brändén and Tooze1991; Fersht, Reference Fersht1998; Liljas et al., Reference Liljas, Liljas, Ash, Lindblom, Nissen and Kjeldgaard2017). Hydrophilic amino acids are polar and readily water-soluble. They include Aspartic acid (D), Glutamic acid (E), Asparagine (N), Glutamine (Q), Lysine (K), Arginine (R), Serine (S), Threonine (T), Histidine (H) and Tyrosine (Y). Conversely, hydrophobic amino acids are nonpolar and water-insoluble. They include Leucine (L), Isoleucine (I), Valine (V), Phenylalanine (F), Methionine (M), Tryptophan (W) and Alanine (A). Cysteine (C) and Glycine (G) are only weakly hydrophilic and hydrophobic, respectively. Proline (P) is a unique case, the nitrogen of Proline is part of the ring structure formed by the side chain and cannot participate in the helical H-bonds. Proline is frequently found at the N-terminus of helices, but if it occurs in the middle of an α-helix, it forces the helix to bend (Liljas et al., Reference Liljas, Liljas, Ash, Lindblom, Nissen and Kjeldgaard2017).
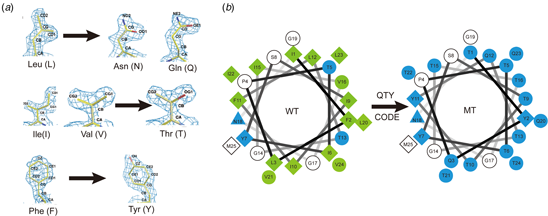
Several amino acids share striking structural similarities within their 1.5 Å-resolution electron density maps, despite their very different chemical properties (Fig. 1). These are: D, N, E and Q versus L; T versus V and I; and Y versus F. The side chains of D, N, E and Q can form four H-bonds with separate water molecules. The carboxylate oxygen atoms of D and E present acceptors for formation of four H-bonds. The amide moieties of N and Q present a donor for formation of two H-bonds (NH2) and an acceptor for formation of two H-bonds (O). The hydroxyl group of S, T and Y can engage in three H-bonds, via two oxygen lone pairs and one O-H bond (Figure S1). Arginine can donate five H-bonds and lysine can donate three H-bonds in the protonated state of their side chains. Histidine has one H-bond donor and one acceptor, and W has one H-bond donor (Figure S1).

Fig. 1. Experimental X-ray electron density maps (~1.5 Å resolution) of 20 amino acids arranged by size. This figure is provided by Dr. Mike Sawaya (UCLA), and used with permission in order to show the individual amino acid electron density maps at high resolution. The density maps demonstrate similar shapes of V and T; L, D, N, E and Q, and F and Y. Please see Dr. Mike Sawaya's original website: http://people.mbi.ucla.edu/sawaya/m230d/Modelbuilding/modelbuilding.html (courtesy of Dr. Michael R. Sawaya of University of California, Los Angeles, CA, USA).
The atomic-resolution molecular structures of proteins unequivocally demonstrate that all 20 natural amino acids are found in α-helices, regardless of their hydrophilic and hydrophobic properties, although some amino acids have a higher propensity to form an α-helix (Chou and Fasman, Reference Chou and Fasman1974; Creighton, Reference Creighton1992).
A strong periodicity in the hydrophobic moment of amino acids in α-helices
David Eisenberg and colleagues in early 1980s had carried out quantitative studies of α-helices using a method they termed the hydrophobic moment (Eisenberg et al., Reference Eisenberg, Weiss and Terwilliger1982; Eisenberg et al., Reference Eisenberg, Weiss and Terwilliger1984). They defined the hydrophobic moment as follows: ‘Periodicities in the polar/apolar character of the amino acid sequence of a protein can be examined by assigning to each residue a numerical hydrophobicity and searching for periodicity in the resulting one-dimensional function. The strength of each periodic component is the quantity that has been termed the hydrophobic moment. When proteins of known three-dimensional structure are examined, it is found that sequences that form a helix tend to have, on average, a strong periodicity in the hydrophobicity of 3.6 residues, the period of the α-helix.’ They analyzed 157 segments of α-helices from globular protein structures from the previous 25 years (1959–1984) and found that the average of these α-helical segments has a strong maximum at 100°. They observed that on average these primary structures are more amphiphilic arranged as α-helices than in other periodic secondary structures (Eisenberg et al., Reference Eisenberg, Weiss and Terwilliger1982; Eisenberg et al., Reference Eisenberg, Weiss and Terwilliger1984). They also pointed out that there are other factors influencing the hydrophobic periodicity, not all segments known to be α-helices give a profile with a maximum at 100°, especially if the hydrophobic residues are not uniformly distributed along one side parallel to the axis but rather arranged in a slanted region across the helix (Eisenberg et al., Reference Eisenberg, Weiss and Terwilliger1982; Eisenberg et al., Reference Eisenberg, Weiss and Terwilliger1984). However, they did not explicitly classify the three chemically distinct α-helix types.
Three chemically distinct α-helix types
The α-helix can be classified into three chemically distinct types (Fig. 2). Although they are significantly different in their chemical properties and water-solubility, they have nearly identical molecular structures, namely: (i) a 1.5 Å rise per amino acid, (ii) a 100° rotation per amino acid, (iii) 3.6 amino acids per helical turn, (iv) a 5.4 Å rise per helical turn, and (v) the key feature, i.e. each NH-group of an amino acid forms an H-bond with the C = O group of the amino acid 4 residues away. The latter, repeated i, i + 4 H-bonding pattern is the most prominent characteristic of an α-helix (Stryer, Reference Stryer1981; Brändén and Tooze, Reference Brändén and Tooze1991; Fersht, Reference Fersht1998; Liljas et al., Reference Liljas, Liljas, Ash, Lindblom, Nissen and Kjeldgaard2017).

Fig. 2. Schematic illustrations of three chemically distinct types of α-helices: Type I hydrophilic, Type II hydrophobic, Type III Janus (% hydrophilic/hydrophobic). The Type I α-helix is mostly comprised of hydrophilic amino acids including Aspartic acid (D), Glutamic acid (E), Asparagine (N), Glutamine (Q), Lysine (K), Arginine (R), Serine (S), Threonine (T), and Tyrosine (Y) that are commonly found on the outer layer in water-soluble globular proteins. The Type II α-helix is mostly comprised of hydrophobic amino acids Leucine (L), Isoleucine (I), Valine (V), Phenylalanine (F), Methionine (M), Tryptophan (W) and Alanine (A) that are commonly found in helical transmembrane segments in membrane proteins. The Type III amphiphilic α-helix is mostly comprised of hydrophilic and hydrophobic amino acids that are partitioned on two faces: hydrophobic face and hydrophilic face.
The Type I α-helix is mostly comprised of hydrophilic amino acids, including D, E, N, Q, K, R, S, T and Y, and is commonly found on the outer layer in water-soluble globular proteins, and in some cases, in the inner layer of membrane helices, away from the lipid bilayer. The Type II α-helix is mostly comprised of hydrophobic amino acids, including L, I, V, F, M, A, W and P, and is commonly found in the helical transmembrane segments of membrane proteins and buried in the interior of water-soluble proteins. The Type III amphiphilic α-helix is almost equally comprised of hydrophilic and hydrophobic amino acids that are sometimes partitioned on the hydrophobic face and the hydrophilic face. Using an analogy, we can think of the two faces as the front and back of our fingers. The Type III α-helix is sometimes attached to the surface of membrane lipid bilayers, or partially buried in the hydrophobic core and partially exposed on the surface of water-soluble globular proteins, or in the integral membrane pores that face away from the hydrophobic lipid bilayer. In the literature glycine (G) is often counted as hydrophobic because its side chain hydrogen does not engage in H-bonds, but is only weakly hydrophobic at the same time. If we were to exclude glycine from the list of hydrophobic amino acids, Type I and Type III α-helices would exhibit different overall percentages of hydrophobicity.
Two classes of proteins: hydrophilic and hydrophobic
Natural proteins can be generally divided into two classes: Class I is hydrophilic and Class II is hydrophobic (Stryer, Reference Stryer1981; Brändén and Tooze, Reference Brändén and Tooze1991; Fersht, Reference Fersht1998; Liljas et al., Reference Liljas, Liljas, Ash, Lindblom, Nissen and Kjeldgaard2017). Class I hydrophilic proteins include water-soluble proteins that reside in the cytoplasm, such as hemoglobin, as well as metabolic enzymes and circulating proteins outside cells, such as growth factors, hormones, and antibodies. Class II hydrophobic proteins comprise the integral membrane proteins that are embedded in cellular and other membranes. They include G protein-coupled receptors (GPCRs), membrane transporters and ion channels as well as the photosynthesis machinery. The Class I proteins are generally water-soluble, and the Class II proteins, as integral membrane proteins, are generally water-insoluble. In order to solubilize Class II proteins, various detergent/surfactants are required after isolating them from their lipid-bilayer membrane environment (Lin and Guidotti, Reference Lin and Guidotti2009; Linke, Reference Linke2009; Duquesne and Sturgis, Reference Duquesne and Sturgis2010).
Long α-helices in proteins
Some proteins are almost entirely made up of α-helices, including trompomyosin (Doran et al., Reference Doran, Pavadai, Rynkiewicz, Walklate, Bullitt, Moore, Regnier, Geeves and Lehman2020) and the helical parts of keratins (Lee et al., Reference Lee, Kim, Li, Leahy and Coulombe2020) (Fig. 3). These proteins are often involved in cytoskeletal structural scaffolds, similar to the scaffolds used to construct buildings. A single α-helix structure of trompomyosin consists of 164 amino acids (~244 Å long, or ~24.4 nm) (Doran et al., Reference Doran, Pavadai, Rynkiewicz, Walklate, Bullitt, Moore, Regnier, Geeves and Lehman2020). In crystal structures determined for portions of keratins, a single α-helix is ~90-amino acids long (Lee et al., Reference Lee, Kim, Li, Leahy and Coulombe2020). It has been suggested that the α-helical parts (~330 aa) of keratins are likely to be as long as ~500 Å (~50 nm) (Hanukoglu and Fuchs, Reference Hanukoglu and Fuchs1983), but currently no molecular structure is available. Another long α-helix is hemagglutinin of influenza virus, which has 52 amino acids (~77 Å) and is crucial for viral fusion with the host membrane to confer entrance into the cell for infection during pH change (Gamblin et al., Reference Gamblin, Haire, Russell, Stevens, Xiao, Ha, Vasisht, Steinhauer, Daniels, Elliot, Wiley and Skehel2004). All 20 amino acids are found in these α-helices, regardless of their chemical properties. In the long 164-amino acid α-helix example of pig trompomyosin, 18 kinds of amino acids are present with only P and W missing (Doran et al., Reference Doran, Pavadai, Rynkiewicz, Walklate, Bullitt, Moore, Regnier, Geeves and Lehman2020). However, in the transmembrane helices of glucose transporter GLUT1 (Custódio et al., Reference Custódio, Paulsen, Frain and Pedersen2021), there are two W and five P residues. Thus, all 20 amino acids are observed in α-helices.

Fig. 3. Long α-helices in proteins. There are some examples of long α-helices in proteins that contain most of the 20 amino acids. (a) Human keratin Type I (K1C14) where the α-helix is made of 90 aa; (b) The α-helix of hemagglutinin of influenza virus has 52 amino acids and is crucial for viral fusion with the host membrane to confer entrance into the cell for infection. (c) The α-helix crystal structure of trompomyosin (TPM1, 4.25 Å) is comprised of 164 aa. (d) Keratin Type II (K2C5) where the α-helix is made of 91 aa. It has been suggested that keratin helices could be as long as 50 nm comprising 330 aa.
Type I α-helix: hydrophilic and highly water-soluble
The Type I hydrophilic α-helix is mostly comprised of hydrophilic amino acids D, E, N, Q, K, R, S, T, Y (Table 1, Fig. 4). G has also been found in Type I α-helices. It is counted as hydrophobic because its ‘side chain’ H atom does not form H-bonds. At the same time this amino acid is only weakly hydrophobic. If G is not counted as hydrophobic, the overall % hydrophobicity of G-containing helices will have to be adjusted.

Fig. 4. Type I α-helix comprises between 73%-82% hydrophilic amino acids (Also see Table 1). (a) Yeast Zuotin α-helix 5 (13/16 = ~81% hydrophilic), (b) Troponin T α-helix 2 (TNNT), (14/17 = 82.2% hydrophilic), (c) Troponin I (TNNI3), α-helix 2, (13/17 = 76.5%), (d) Troponin T α-helix 1, (6/22 = 73% hydrophilic.
Table 1. Three chemically distinct types of α-helices: hydrophilic, hydrophobic and amphiphilic

The α-helix can be classified into three chemically distinct Types. The Type I hydrophilic α-helix is mostly comprised of hydrophilic amino acids D, E, N, Q, K, R, S, T, Y; Type II hydrophobic α-helix is mostly comprised of hydrophobic amino acids L, I, V, F, M, P and A; Type III amphiphilic alpha-helix is comprised of both hydrophilic and hydrophobic amino acids, the hydrophobic face and the hydrophilic face. The Type III α-helix is sometimes attached to the surface of the membrane lipid bilayer, or partially buried in the hydrophobic core and partially exposed on the surface of water-soluble globular proteins. Glycine (G) is counted as hydrophobic because its side chain does not engage in H-bonding, although it is only very weakly hydrophobic. If G is not counted as hydrophobic, the percentages will be different.
It is difficult to find long Type I α-helices with only hydrophilic residues. However, it is common to find the Type I α-helix in water-soluble proteins. Figure 4 shows Type I α-helices from proteins with between 73-82% hydrophilic amino acids (Table 1). For example, (i) yeast Zuotin α-helix H5: 13/16 = 81.3% hydrophilic (Zhang et al., Reference Zhang, Lockshin, Herbert, Winter and Rich1992; Lee et al., Reference Lee, Sharma, Shrestha, Bingman and Craig2016); (ii) Troponin T (TNNT) α-helix 2: 14/17 = 82.2% hydrophilic (Takeda et al., Reference Takeda, Yamashita, Maeda and Maeda2003); (iii) Troponin I (TNNI3) α-helix 2: 13/17 = 76.5% hydrophilic (Yaguchi et al., Reference Yaguchi, Yaguchi and Tanaka2017); (iv) Troponin T (TNNT) α-helix 1: 16/22 = 72.7% hydrophilic (Takeda et al., Reference Takeda, Yamashita, Maeda and Maeda2003). It is clear that countless Type I α-helices exist in proteins.
Type II α-helix: hydrophobic and spanning membranes
The Type II hydrophobic α-helix is mostly comprised of hydrophobic amino acids L, I, V, F, M, P, W and A (Fig. 5 and Table 1). Therefore, Type II α-helices are mostly found embedded in lipid bilayer membranes. They constitute transmembrane α-helices that contain between 81% and 91% hydrophobic amino acids. For example, (i) GLUT1 (TM9): 21/25 = 84% hydrophobic (Custódio et al., Reference Custódio, Paulsen, Frain and Pedersen2021); (ii) GLUT3: 16/18 = 88.9% hydrophobic (Deng et al., Reference Deng, Sun, Yan, Ke, Jiang, Xiong, Ren, Hirata, Yamamoto, Fan and Yan2015); (iii) pufL, L-chain (TM1): 22/24 = 91.6% hydrophobic (Xu et al., Reference Xu, Axelrod, Abresch, Paddock, Okamura and Feher2004); (iv) pufM, M-chain (TM5): 18/22 = 81.8% hydrophobic (Xu et al., Reference Xu, Axelrod, Abresch, Paddock, Okamura and Feher2004); e) CCR5 (TM1): 25/31 = ~81% hydrophobic (Tan et al., Reference Tan, Zhu, Li, Chen, Han, Kufareva, Li, Ma, Fenalti, Li, Zhang, Xie, Yang, Jiang, Cherezov, Liu, Stevens, Zhao and Wu2013). Again, it is uncommon to find a completely hydrophobic α-helix, even in transmembrane domains. Other Type II α-helices are also found in interior domains of water-soluble proteins, i.e. in the hydrophobic core.

Fig. 5. Type II α-helix is a transmembrane helix comprising 81%-91% hydrophobic amino acids. (a) GLUT1 (TM9) 21/25 = 85% hydrophobic, (b) GLUT3, 16/18 = 89% hydrophobic, (c) pufL, L-chain (TM1) (22/24 = 91.6% hydrophobic), (d) pufM, M-chain (TM5), 18/22 = (82%) hydrophobic, (e) CCR5 (TM1), (25/31 = ~81% hydrophobic).
Type III α-helix: amphiphilic
The Type III amphiphilic α-helix is comprised of almost equal numbers of hydrophilic and hydrophobic amino acids that are partitioned on hydrophobic and hydrophilic faces (Fig. 6). The Type III α-helix is sometimes attached to the surface of lipid bilayer membranes, or partially buried in the hydrophobic core and partially exposed on the surface of water-soluble globular proteins (Stryer, Reference Stryer1981; Brändén and Tooze, Reference Brändén and Tooze1991; Fersht, Reference Fersht1998; Liljas et al., Reference Liljas, Liljas, Ash, Lindblom, Nissen and Kjeldgaard2017). Examples include α-helices in, (i) human hemoglobin beta subunit, 50% hydrophilic/hydrophobic amino acids, (ii) T4 lysozyme with 47.4% hydrophilic and 52.6% hydrophobic amino acids (Mooers and Matthews, Reference Mooers and Matthews2004), (iii) alcohol dehydrogenase with 53.8% hydrophilic and 46.2% hydrophobic amino acids (Niederhut et al., Reference Niederhut, Gibbons, Perez-Miller and Hurley2001), (iv) Cytochrome b562, a coiled-coil tetramer (H4) with 46% hydrophilic and 54% hydrophobic amino acids (Lederer et al., Reference Lederer, Glatigny, Bethge, Bellamy and Matthew1981), and (v) the designed 29-amino acids, trimeric coiled-coil VALD with 51.7% hydrophilic and 48.3% hydrophobic amino acids (Ogihara et al., Reference Ogihara, Weiss, Degrado and Eisenberg1997). There are also many Type III α-helices in proteins that reside on the surfaces of membranes, whereby one side interacts with the hydrophobic lipids from the membrane and the other is exposed to the aqueous environment.

Fig. 6. Type III α-helix is an amphiphilic helix and comprises both hydrophilic and hydrophobic amino acids. (a) T4 lysozyme with 47.4% hydrophilic and 52.6% hydrophobic amino acids, (b) alcohol dehydrogenase with 53.8% hydrophilic and 46.2% hydrophobic amino acids, (c) Cytochrome b562 is a coiled-coil tetramer (H4) with 46% hydrophilic and 54% hydrophobic amino acids, (d) Designed 29 aa trimeric coiled-coil VALD (1COI, 2.10 Å) with 51.7% hydrophilic and 48.3% hydrophobic amino acids.
Conversion of a hydrophobic α-helix to a hydrophilic α-helix using a simple QTY code
Over a decade ago, in 2010, Alexander Rich posed a question to one of us (S.Z.) ‘Can you convert a hydrophobic α-helix to a hydrophilic one?’ which prompted the immediate answer ‘Yes, because hemoglobin is composed mostly of α-helices and it is one of the most water-soluble proteins’ But how? Several previous attempts were not successful, e.g. (Mitra et al., Reference Mitra, Steitz and Engelman2002). Others succeeded by using sophisticated approaches, including computer-assisted modeling, simulations and introduction of specific mutations in a given protein (Slovic et al., Reference Slovic, Summa, Lear and DeGrado2003, Reference Slovic, Kono, Lear, Saven and DeGrado2004, Reference Slovic, Stayrook, North and Degrado2005; Zhang SQ et al., Reference Zhang, Tao, Qing, Tang, Skuhersky, Corin, Tegler, Wassie, Wassie, Kwon, Suter, Entzian, Schubert, Yang, Labahn, Kubicek and Maertens2018a; Zhang S et al., Reference Zhang, Huang, Yang, Kratochvil, Lolicato, Liu, Shu, Liu and DeGrado2018b). For each water-insoluble protein, it is necessary to go through a rigorous and time-consuming exercise to render it water-soluble. There was no straightforward and shared code to simplify such hydrophobic to hydrophilic protein conversion.
A simple QTY code (Fig. 7) was devised by considering the 1.5 Å-resolution electron density maps of the 20 amino acids (Fig. 1). It is clear that several hydrophobic amino acids closely resemble those of hydrophilic amino acids in terms of shape (Fig. 1). Thus, the QTY code is based on the fact that the electron density (computed at sufficient resolution) of the hydrophobic L is similar to that of the hydrophilic N and Q. The electron density of the hydrophobic I and V are similar to that of the hydrophilic T. Lastly, the electron density of the hydrophobic F is similar to that of the hydrophilic Y (Fig. 1). Although water molecules also form H-bonds with D (- charge), E (−), K (+) and R (+), these residues introduce charge, thereby altering the surface property of proteins. Thus, they are not considered in the QTY code. Since the sidechain of N is proximate to the backbone of the polypeptide chain and sometimes interferes with H-bonds to the amide moiety, and often maps to turns, it was not included in the code.
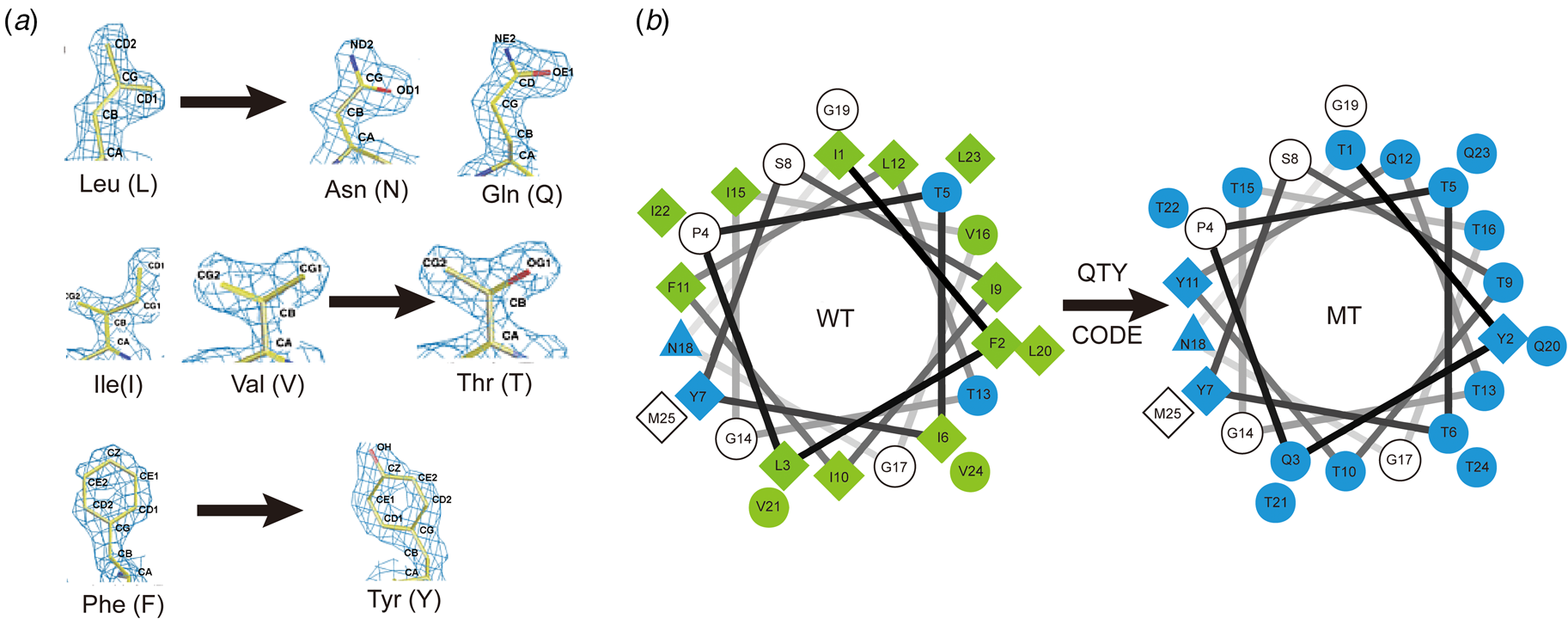
Fig. 7. The QTY code and how it replaces L, V, I and F with Q, T and Y. (A) Crystallographic electron density maps of the following amino acids: Leucine (L), Asparagine (N), Glutamine (Q), Isoleucine (I), Valine (V), Threonine (T), Phenylalanine (F) and Tyrosine (Y). The density maps of L, N and Q are very similar. Likewise, the density maps of I, V and T are similar, and the density maps of F and Y are similar. The side chains of L, V, I, and F cannot form any H-bonds with water, thus rendering them water-insoluble. On the other hand, N and Q can form four H-bonds with four water molecules, two as H-bond donors and two as H-bond acceptors (SI Appendix Fig. S7). Likewise, three water molecules can form H-bonds with the –OH (two H-bond donors and one H-bond acceptor) of Thr (T) and Tyr (Y). Both L and Q have high tendencies to form α-helices, but N frequently occurs at turns. Thus, Q was used to replace L, but not N. I, V and T are all β-branched amino acids and their density maps are very similar, indicating similar shapes. (B) Helical wheels before and after applying the QTY code to transmembrane helical segment 1 (TM1) of CXCR4. Amino acids that interact with water molecules are light blue in color. The QTY code conversions render the α-helical segment water-soluble.
The QTY code was initially applied to convert transmembrane (TM) α-helices of seven G protein-coupled receptors (GPCRs), namely the chemokine receptors CCR5, CCR9, CCR10, CXCR2, CXCR4, CXCR5, CXCR7 (Zhang et al., Reference Zhang, Tao, Qing, Tang, Skuhersky, Corin, Tegler, Wassie, Wassie, Kwon, Suter, Entzian, Schubert, Yang, Labahn, Kubicek and Maertens2018a, Reference Zhang, Huang, Yang, Kratochvil, Lolicato, Liu, Shu, Liu and DeGrado2018b; Qing et al., Reference Qing, Han, Fei, Skuhersky, Badr, Chung, Schubert and Zhang2019; Hao et al., Reference Hao, Jin, Zhang and Qing2020; Tegler et al., Reference Tegler, Corin, Skuhersky, Pick, Vogel and Zhang2020; Skuhersky et al., Reference Skuhersky, Tao, Qing, Smorodina, Jin and Zhang2021; Tao et al., Reference Tao, Tang, Zhang, Li and Xu2022). Despite ~20–30% overall amino acid changes (46–58% transmembrane domain amino acid changes), these water-soluble QTY variant receptors still retained their overall structure, with a similar amount of α-helical content, and folded into stable proteins (Fig. 8). Most importantly, they were able to bind their respective ligands, with affinities that were similar to those of the natural receptors. After using the QTY code to replace the hydrophobic amino acids L, I, V and F in the transmembrane domain, the hydrophobicity on the surface of membrane proteins is reduced significantly. They in turn became more hydrophilic for several GPCRs, including CCR5, CCR9, CXC2 and CXCR4 (Fig. 8). Currently, experimental determinations of the structures of QTY variant receptors are in progress in collaborations with other laboratories.

Fig. 8. Surface hydrophobic patches of X-ray crystal structures of native chemokine receptors and AlphaFold2 predicted water-soluble QTY variants. The native GPCR receptors mostly expose hydrophobic residues leucine (L), isoleucine (I), valine (V) and phenylalanine (F) to the hydrophobic lipid bilayer of the cell membrane. After replacing L, I, V, F with polar amino acids, glutamine (Q), threonine (T) and tyrosine (Y), the surfaces are much less hydrophobic. The large surface hydrophobic patch (yellow color) of the native receptors determined by X-ray crystallography: (a) CCR5, (b) CCR9, (c) CXCR2 and (d) CXCR4. The hydrophobic patch is significantly reduced on the transmembrane domains for the AlphaFold2 predicted water-soluble QTY variants: (e) CCR5QTY, (f) CCR9QTY, (g) CXCR2QTY, (h) CXCR4QTY. These QTY variants become water-soluble without any detergent. The N- and C-termini are removed for clarity.
We later successfully applied the QTY code to four cytokine receptors interleukins IL4Rα, IL10Rα and interferon INFγR1 and INFλR1 that have a single transmembrane α-helix (Hao et al., Reference Hao, Jin, Zhang and Qing2020). Other laboratories also independently reproduced the water-soluble QTY variant receptors, carried out ligand binding experiments and obtained similar results. Recently, a water-soluble CCR10QTY variant has been used as antigen to generate monoclonal antibodies.
Currently, we are interested in studying glucose transporters (12TM) since they are directly involved in proliferation and metastasis of a wide spectrum of cancer cells that constantly demand energy supply (Barron et al., Reference Barron, Bilan, Tsakiridis and Tsiani2016). The QTY code has also been applied to design water-soluble glucose transporter QTY variants (Fig. 9). Despite significant amino acid changes (overall >24% and TM >44%), the crystal structure of GLUT1 and the predicted structure of its QTY variant GLUT1QTY superpose well (RMSD = 1.55 Å). Likewise, the crystal structure of GLUT3 closely resembles that of the predicted structure of its variant GLUT3QTY (RMSD = 1.03 Å) (Smorodina et al., Reference Smorodina, Tao, Qing, Jin, Yang and Zhang2022). We believe that this simple QTY code could be widely applicable to diverse membrane proteins that have TM α-helices.

Fig. 9. Superimpositions of two glucose transporters GLUT1 and GLUT3 and their AlphaFold2 predicted QTY water-soluble variants. For each superimposition, the structures are shown in the side view (left), and the top view (right). The X-ray crystal structures of natural GLU1 (6THA, 2.4 Å), GLUT3 (4ZW9, 1.5 Å). (a and b) The crystal structure of native GLUT1 (magenta) is overlaid on the AlphaFold2 predicted water-soluble variant GLUT1QTY (cyan). The RMSD is 1.55 Å for GLUT1 and GLUT1QTY. (c and d) The GLUT3 CryoEM structure (magenta) is overlaid on the AlphaFold2 predicted water-soluble variant GLUT3QTY (cyan). The RMSD is 1.03 Å for GLUT3 and GLUT3QTY. For clarity, long N- and C-termini are removed.
Leonardo da Vinci remarked that ‘Simplicity is the ultimate sophistication’. Such a simple QTY code may stimulate researchers to systematically convert water-insoluble or aggregated proteins into water-soluble variants, not only to further protein structural studies, but also for biotechnological and nanotechnological applications and beyond.
Two simple molecular codes
Complementary DNA base pairing is governed by a simple code: A pairs with T and G pairs with C and vice versa (Fig. 10). Once the sequence of one DNA strand is identified, that of its complement is automatically known. When the right-handed model of the DNA double helix was first discovered in the early 1950s, based on X-ray fiber diffraction experiments and base-pairing considerations, it was thought it would exhibit limited conformational variation beyond the two forms apparent in fiber diffraction images (Frank-Kamenetskii, Reference Frank-Kamenetskii1993). Through systematically studying single-crystal structures of DNA alone and in complex with proteins at high resolution, it was revealed that the double helix is not uniform at all (Rich, Reference Rich1983; Frank-Kamenetskii, Reference Frank-Kamenetskii1993). Thus, DNA not only forms a left-handed double helix (Z-form) (Wang et al., Reference Wang, Quigley, Kolpak, Crawford, van Boom, van der Marel and Rich1979), but it also adopts a wide range of conformational variants, including A-form, B-form, C-form, D-form, E-form, H-form, writhing and kinked species, and more (Egli, Reference Egli, Blackburn, Egli, Gait and Watts2022). In retrospect, it is not surprising that DNA structure exhibits many variations, consistent with its diverse biological activities and functions in cells.

Fig. 10. Two simple codes. (a) DNA code: base-pairing specificity and complementarity. The DNA code is bi-directional and reversible. (b) QTY code: matching shapes of hydrophobic and hydrophilic amino acid side chains. The QTY code is also bi-directional and reversible.
The three chemical types of α-helix, although known to structural biologists (Eisenberg et al., Reference Eisenberg, Weiss and Terwilliger1982, Reference Eisenberg, Weiss and Terwilliger1984), are much less known to non-structural biologists and non-protein scientists. We here explicitly classify three chemically distinct α-helix types to put a spotlight on them. This classification will likely provide us with an opportunity to fully understand subtle variations in the three chemical types of α-helix in high-resolution structures, especially when combined with the results of artificial intelligence and machine learning tools. We believe that the explicit classification of the three chemically distinct types of α-helix combined with the simple QTY code (Fig. 10) will likely stimulate scientists not only to further study protein structure and function, but also use the concept for protein design.
Supplementary material
The supplementary material for this article can be found at https://doi.org/10.1017/S0033583522000063.














 Open access
Open access