



No CrossRef data available.
Article contents
The crystal structure of charmarite – the first case of a 11 × 11 Å superstructure mesh in layered double hydroxides
Published online by Cambridge University Press: 08 March 2024
Abstract
Charmarite, Mn4Al2(OH)12CO3⋅3H2O, is a hydrotalcite supergroup member (or layered double hydroxide, LDH) with a previously unknown crystal structure and a Mn2+-analogue of quintinite (commonly erroneously reported as ‘2:1 hydrotalcite’). The single-crystal X-ray diffraction (XRD) data were obtained from the specimen from Mont Saint-Hilaire, Québec, Canada and are best processed in the space group P$\bar{3}$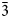 , a = 10.9630(4), c = 15.0732(5) Å and V = 1568.89(12) Å3. The crystal structure has been solved by direct methods and refined to R1 = 0.0750 for 3801 unique reflections with Fo > 2σ(Fo). The charmarite structure has long-range periodicity in the xy plane due to $2\sqrt 3$
, a = 10.9630(4), c = 15.0732(5) Å and V = 1568.89(12) Å3. The crystal structure has been solved by direct methods and refined to R1 = 0.0750 for 3801 unique reflections with Fo > 2σ(Fo). The charmarite structure has long-range periodicity in the xy plane due to $2\sqrt 3$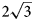 a’ × $2\sqrt 3$
a’ × $2\sqrt 3$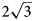 a’ scheme (or 11 × 11 Å) determined for LDHs for the first time (where a’ is a subcell parameter ≈ 3.2 Å). This periodicity is produced by the combination of two superstructures formed by: (1) Mn2+ and Al3+ ordering in the metal-hydroxide layers [Mn4Al2(OH)12]2+ according to the $\sqrt 3$
a’ scheme (or 11 × 11 Å) determined for LDHs for the first time (where a’ is a subcell parameter ≈ 3.2 Å). This periodicity is produced by the combination of two superstructures formed by: (1) Mn2+ and Al3+ ordering in the metal-hydroxide layers [Mn4Al2(OH)12]2+ according to the $\sqrt 3$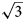 a’ × $\sqrt 3$
a’ × $\sqrt 3$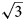 a’ pattern and (2) the (CO3)2– ordering according to the 2a’ × 2a’ pattern in the [CO3(H2O)3]2– interlayer sheet in order to avoid close contacts between adjacent carbonate groups. The $2\sqrt 3$
a’ pattern and (2) the (CO3)2– ordering according to the 2a’ × 2a’ pattern in the [CO3(H2O)3]2– interlayer sheet in order to avoid close contacts between adjacent carbonate groups. The $2\sqrt 3$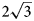 a’ × $2\sqrt 3$
a’ × $2\sqrt 3$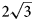 a’ superstructure is an example of the adaptability of the components of the interlayer space to the charge distribution of the metal-hydroxyl layers. The Mn2+ and Al3+ cations have a large difference in size, which apparently leads to the considerable degree of their order as di- and trivalent cations resulting in a higher degree of statistical order of the interlayer components. Both powder and single-crystal XRD data show that the samples studied belong to the hexagonal branch of two-layer polytypes (2T or 2H) with d00n ≈ 7.57 Å. The chemical composition of the samples studied is close to the ideal formula. The Raman spectrum of charmarite is reported and the band assignment is provided.
a’ superstructure is an example of the adaptability of the components of the interlayer space to the charge distribution of the metal-hydroxyl layers. The Mn2+ and Al3+ cations have a large difference in size, which apparently leads to the considerable degree of their order as di- and trivalent cations resulting in a higher degree of statistical order of the interlayer components. Both powder and single-crystal XRD data show that the samples studied belong to the hexagonal branch of two-layer polytypes (2T or 2H) with d00n ≈ 7.57 Å. The chemical composition of the samples studied is close to the ideal formula. The Raman spectrum of charmarite is reported and the band assignment is provided.
Keywords
- Type
- Article
- Information
- Copyright
- Copyright © The Author(s), 2024. Published by Cambridge University Press on behalf of The Mineralogical Society of the United Kingdom and Ireland
Footnotes
Associate Editor: G. Diego Gatta
References
Andersen, A.B.A., Henriksen, C., Wang, Q., Ravnsbæk, D.B., Hansen, L.P. and Nielsen, U.G. (2021) Synthesis and thermal degradation of MAl4(OH)12SO4⋅3H2O with M = Co2+, Ni2+, Cu2+, and Zn2+. Inorganic Chemistry, 60, 16700–16712.CrossRefGoogle Scholar
Arakcheeva, A.V., Pushcharovskii, D.Y., Rastsvetaeva, R.K., Atencio, D. and Lubman, G.U. (1996) Crystal structure and comparative crystal chemistry of Al2Mg4(OH)12(CO3)⋅3H2O, a new mineral from the hydrotalcite-manasseite group. Crystallography Reports, 41, 972–981.Google Scholar
Bonaccorsi, E., Merlino, S. and Orlandi, P. (2007) Zincalstibite, a new mineral, and cualstibite: crystal chemical and structural relationships. American Mineralogist, 92, 198–203.10.2138/am.2007.2240CrossRefGoogle Scholar
Bosa, G., Silva, C., Brito, B., Terzi, C., Wypych, F. and Nakagaki, S. (2023) Revisiting the synthesis of gibbsite, a precursor for Li/Al LDH synthesis and its use as a support for porphyrin immobilization. Journal of the Brazilian Chemical Society, 35, https://doi.org/10.21577/0103-5053.20230095.Google Scholar
Britto, S. and Kamath, P.V. (2011) Polytypism in the lithium–aluminum layered double hydroxides: The [LiAl2(OH)6]+ layer as a structural synthon. Inorganic Chemistry, 50, 5619–5627.10.1021/ic200312gCrossRefGoogle ScholarPubMed
Britvin, S.N., Dolivo-Dobrovolsky, D.V. and Krzhizhanovskaya, M.G. (2017) Software for processing the X-ray powder diffraction data obtained from the curved image plate detector of Rigaku RAXIS Rapid II diffractometer. Zapiski Rossiiskogo Mineralogicheskogo Obshchestva, 146, 104–107 [in Russian with English abstract].Google Scholar
Bruker-AXS (2009) Topas, version 4.2; General Profile and Structure Analysis Software for Powder Diffraction Data. Bruker-AXS, Karlsruhe, Germany.Google Scholar
Chao, G.Y. and Gault, R.A. (1997) Quintinite-2H, quintinite-3T, charmarite-2H, charmarite-3T and caresite-3T, a new group of carbonate minerals related to the hydrotalcite–manasseite group. The Canadian Mineralogist, 35, 1541–1549.Google Scholar
Dolomanov, O.V., Blake, A.J., Champness, N.R. and Schröder, M. (2009) OLEX: new software for visualization and analysis of extended crystal structures. Journal of Applied Crystallography, 36, 1283–1284.10.1107/S0021889803015267CrossRefGoogle Scholar
Evans, D.G. and Slade, R.C.T. (2005) Structural aspects of layered double hydroxides. Pp. 1–87 in: Layered Double Hydroxides (Duan, X. and Evans, D.G. editors). Structure and Bonding, vol 119. Springer, Berlin, Heidelberg.Google ScholarPubMed
Forano, C., Hibino, T., Leroux, F. and Taviot-Guého, C. (2006) Chapter 13.1 layered double hydroxides. Developments in Clay Science, 1, 1021–1095.10.1016/S1572-4352(05)01039-1CrossRefGoogle Scholar
Grand, L.M., Palmer, S.J. and Frost, R.L. (2010) Synthesis and thermal stability of hydrotalcites containing manganese. Journal of Thermal Analysis and Calorimetry, 100, 981–985.CrossRefGoogle Scholar
Guilherme, V.A., Cunha, V.R.R., de Paula, E., de Araujo, D.R. and Constantino, V.R.L. (2022) Anti-inflammatory and analgesic evaluation of a phytochemical intercalated into layered double hydroxide. Pharmaceutics, 14, 934.10.3390/pharmaceutics14050934CrossRefGoogle ScholarPubMed
Karim, A.V., Hassani, A., Eghbali, P. and Nidheesh, P.V. (2022) Nanostructured modified layered double hydroxides (LDHs)-based catalysts: a review on synthesis, characterization, and applications in water remediation by advanced oxidation processes. Current Opinion in Solid State and Materials Science, 26, 100965.CrossRefGoogle Scholar
Karpenko, V.Yu., Zhitova, E.S., Pautov, L.A., Agakhanov, A.A., Siidra, O.I., Krzhizhanovskaya, M.G., Rassulov, V.A. and Bocharov, V.N. (2020) Akopovaite, Li2Al4(OH)12(CO3)(H2O)3, a new Li member of the hydrotalcite supergroup from Turkestan Range, Kyrgyzstan. Mineralogical Magazine, 84, 301–311.10.1180/mgm.2020.10CrossRefGoogle Scholar
Krivovichev, S.V., Yakovenchuk, V.N., Zhitova, E.S., Zolotarev, A.A., Pakhomovsky, Y.A. and Ivanyuk, G.Yu. (2010a) Crystal chemistry of natural layered double hydroxides. I. Quintinite 2H–3c from the Kovdor alkaline massif, Kola peninsula, Russia. Mineralogical Magazine, 74, 821–832.CrossRefGoogle Scholar
Krivovichev, S.V., Yakovenchuk, V.N., Zhitova, E.S., Zolotarev, A.A., Pakhomovsky, Y.A. and Ivanyuk, G.Yu. (2010b) Crystal chemistry of natural layered double hydroxides. 2. Quintinite–1M: first evidence of a monoclinic polytype in M 2+-M 3+ layered double hydroxides. Mineralogical Magazine, 74, 833–840.CrossRefGoogle Scholar
Krivovichev, S.V., Antonov, A.A., Zhitova, E.S., Zolotarev, A.A., Krivovichev, V.G. and Yakovenchuk, V.N. (2012) Quintinite-1M from the Bazhenov deposit (Middle Urals, Russia): crystal structure and properties. Bulletin of St. Petersburg University. Series 7: Geology, Geography, 2, 3–10 [in Russian with English abstract].Google Scholar
Lafuente, B., Downs, R.T., Yang, H. and Stone, N. (2015) The power of databases: the RRUFF project. Pp. 1–30 in: Highlights in Mineralogical Crystallography (Armbruster, T. and Danisi, R.M., editors). W. De Gruyter, Berlin, Germany.Google Scholar
Mills, S.J., Christy, A.G., Génin, J.-M.R., Kameda, T. and Colombo, F. (2012) Nomenclature of the hydrotalcite supergroup: natural layered double hydroxides. Mineralogical Magazine, 76, 1289–1336.CrossRefGoogle Scholar
Mills, S.J., Christy, A.G. and Schmitt, R.T. (2016) The creation of neotypes for hydrotalcite. Mineralogical Magazine, 80, 1023–1029.10.1180/minmag.2016.080.040CrossRefGoogle Scholar
Momma, K. and Izumi, F. (2011) VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. Journal of Applied Crystallography, 44, 1272–1276.CrossRefGoogle Scholar
Rigaku, (2018) PDXL: Integrated X-Ray Powder Diffraction Software, Version 2.8.4.0 (October 23, 2018). Tokyo, Japan.Google Scholar
Rives, V. (2001) Layered Double Hydroxides: Present and Future. Nova Science Publishers: New York, USA.Google Scholar
Robinson, K., Gibbs, G.V. and Ribbe, P.H. (1971) Quadratic elongation: a quantitative measure of distortion in coordination polyhedra. Science, 172, 567–570.CrossRefGoogle ScholarPubMed
Serna, C.J. (1982) Crystal-chemical study of layered [Al2Li(OH)6]+X−⋅nH2O. Clays and Clay Minerals, 30, 180–184.CrossRefGoogle Scholar
Shannon, R.D. (1976) Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallographica, A32, 751–767.10.1107/S0567739476001551CrossRefGoogle Scholar
Sheldrick, G.M. (2008) A short history of SHELX. Acta Crystallographica, A64, 112–122.CrossRefGoogle Scholar
Sheldrick, G.M. (2015) Crystal structure refinement with SHELXL. Acta Crystallographica, C71, 3–8.Google Scholar
Sissoko, I., Iyagba, E.T., Sahai, R. and Biloen, P. (1985) Anion intercalation and exchange in Al(OH)3 - derived compounds. Journal of Solid State Chemistry, 60, 283–288.CrossRefGoogle Scholar
Sotiles, A.R. and Wypych, F. (2019) Converting Mn/Al layered double hydroxide anion exchangers into cation exchangers by topotactic reactions using alkali metal sulfate solutions. Chemical Communications, 55, 7824–7827.CrossRefGoogle ScholarPubMed
Spek, A.L.J. (2003) Single-crystal structure validation with the program PLATON. Journal of Applied Crystallography, 36, 7–13.10.1107/S0021889802022112CrossRefGoogle Scholar
Theiss, F., López, A., Frost, R.L. and Scholz, R. (2015) Spectroscopic characterisation of the LDH mineral quintinite Mg4Al2(OH)12CO3⋅3H2O. Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy, 150, 758–764.CrossRefGoogle Scholar
Walenta, K. (1984) Cualstibit, ein neues Sekundärmineral aus der Grube Clara im mittleren Schwarzwald (BRD). Chemie der Erde, 43, 255–260.Google Scholar
Wang, S.L., Liu, C.H., Wang, M.K., Chuang, Y.H. and Chiang, P.N. (2009) Arsenate adsorption by Mg/Al–NO3 layered double hydroxides with varying the Mg/Al ratio. Applied Clay Science, 43, 79–85.CrossRefGoogle Scholar
Xu, M. and Wei, M. (2018) Layered double hydroxide-based catalysts: recent advances in preparation, structure, and applications. Advanced Functional Materials, 28, 1802943.CrossRefGoogle Scholar
Zhitova, E.S., Yakovenchuk, V.N., Krivovichev, S. V., Zolotarev, A.A., Pakhomovsky, Y.A. and Ivanyuk, G.Yu. (2010) Crystal chemistry of natural layered double hydroxides. 3. The crystal structure of Mg,Al-disordered quintinite-2H. Mineralogical Magazine, 74, 841–848.10.1180/minmag.2010.074.5.841CrossRefGoogle Scholar
Zhitova, E.S., Krivovichev, S.V., Pekov, I.V., Yakovenchuk, V.N. and Pakhomovsky, Y.A. (2016) Correlation between the d-value and the M2+:M3+ cation ratio in Mg–Al–CO3 layered double hydroxides. Applied Clay Science, 130, 2–11.CrossRefGoogle Scholar
Zhitova, E.S., Popov, M.P., Krivovichev, S.V., Zaitsev, A.N. and Vlasenko, N.S. (2017) Quintinite–1M from the Mariinsky deposit, Ural Emerald Mines, Central Urals, Russia. Geology of Ore Deposits, 59, 745–751.CrossRefGoogle Scholar
Zhitova, E.S., Krivovichev, S.V., Yakovenchuk, V.N., Ivanyuk, G.Yu., Pakhomovsky, Y.A. and Mikhailova, J.A. (2018) Crystal chemistry of natural layered double hydroxides: 4. Crystal structures and evolution of structural complexity of quintinite polytypes from the Kovdor alkaline-ultrabasic massif, Kola peninsula, Russia. Mineralogical Magazine, 82, 329–346.CrossRefGoogle Scholar
Zhitova, E.S., Krivovichev, S.V., Pekov, I.V. and Greenwell, H.C. (2019a) Crystal chemistry of natural layered double hydroxides. 5. Single-crystal structure refinement of hydrotalcite, [Mg6Al2(OH)16](CO3)(H2O)4. Mineralogical Magazine, 83, 269–280.CrossRefGoogle Scholar
Zhitova, E.S., Krivovichev, S.V., Pekov, I.V. and Yapaskurt, V.O. (2019b) Crystal chemistry of chlormagaluminite, Mg4Al2(OH)12Cl2(H2O)2, a natural layered double hydroxide. Minerals, 9, 221.CrossRefGoogle Scholar
Zhitova, E.S., Pekov, I.V., Chaikovskiy, I.I., Chirkova, E.P., Yapaskurt, V.O., Bychkova, Y.V., Belakovsky, D.I., Chukanov, N.V., Zubkova, N.V., Krivovichev, S.V. and Bocharov, V. N. (2019c) Dritsite, Li2Al4(OH)12Cl2⋅3H2O, a new gibbsite-based hydrotalcite supergroup mineral. Minerals, 9, 492.10.3390/min9080492CrossRefGoogle Scholar
Zhitova, E.S., Sheveleva, R.M., Zolotarev, A.A. and Krivovichev, S.V. (2023a) The crystal structure of Mg–Al–CO3 layered double hydroxide. Crystals, 13, 839.CrossRefGoogle Scholar
Zhitova, E.S., Chukanov, N.V., Pekov, I.V., Zolotarev, A.A., Shilovskikh, V.V. and Bocharov, V.N. (2023b) Crystal chemistry of iowaite, Mg6FeIII2(OH)16Cl2×4H2O, a natural layered double hydroxide. Applied Clay Science, 243, 107070.CrossRefGoogle Scholar

Zhitova et al. supplementary material
Zhitova et al. supplementary material

File
571.9 KB