


The increased incidence of obesity and its medical consequences, such as CVD, diabetes and hepatic steatosis, supports the need for the development of strategies for obesity prevention(Reference Bray and Bellanger1). A new paradigm for obesity prevention has emerged in recent years, which has evolved from the notion that nutritional and other environmental factors in early life can have a profound influence on lifelong health(Reference Barker2–Reference Vickers, Breier and Cutfield4). In particular, a key role for leptin, a component of breast milk, has been unveiled(Reference Palou and Pico5) and its potential beneficial impact on obesity and metabolic disease has been highlighted recently(Reference Jones6).
The anorexigenic hormone leptin has been identified recently as a factor provided to the suckling infant during lactation, which may help prevent obesity and other metabolic alterations later in life(Reference Palou and Pico5). Leptin is present in maternal milk(Reference Casabiell, Pineiro and Tome7, Reference Houseknecht, McGuire and Portocarrero8), but not in infant formula(Reference O'Connor, Funanage and Locke9), and levels in milk are correlated with maternal BMI (or adiposity) and with plasma leptin concentration(Reference Houseknecht, McGuire and Portocarrero8, Reference Miralles, Sanchez and Palou10). Results of a study in non-obese women, who breast-fed their infants for at least 6 months, have indicated that infant body weight during the first 2 years may be influenced by milk leptin concentration during the first stages of lactation(Reference Miralles, Sanchez and Palou10), suggesting that milk-borne maternal leptin may provide moderate protection to infants from excessive weight gain. A cause–effect relationship has been demonstrated in neonatal rats that were supplemented with physiological doses of leptin during the suckling period, and were subsequently protected against the development of overweight/obesity in adulthood(Reference Pico, Oliver and Sanchez11, Reference Sanchez, Priego and Palou12). Importantly, these animals were more resistant to dietary obesity induced by feeding a high-fat (HF) diet(Reference Pico, Oliver and Sanchez11, Reference Priego, Sanchez and Palou13).
The lower body-weight gain of leptin-treated animals was explained, at least partially, by lower food intake and enhanced sensitivity to leptin(Reference Pico, Oliver and Sanchez11, Reference Sanchez, Priego and Palou12). Changes in the hypothalamic expression of factors involved in the regulation of food intake were observed, particularly in animals exposed to a HF diet(Reference Pico, Oliver and Sanchez11). Under these dietary conditions, leptin-treated animals showed higher expression levels of the main anorexigenic neuropeptide, pro-opiomelanocortin (POMC), while this increase was not found in control animals(Reference Pico, Oliver and Sanchez11).
The expression of POMC is centrally regulated by circulating leptin and insulin(Reference Schwartz, Woods and Porte14). Chronic exposure to a HF diet has been associated with dysregulation of hypothalamic circuits, particularly impairment of the anorexigenic leptin–POMC system(Reference Priego, Sanchez and Pico15), including functional resistance to insulin and leptin, which is associated with overfeeding and overweight(Reference Pico, Oliver and Sanchez11). Of interest, animals that were supplemented with leptin during the suckling period were resistant to the decrease in hypothalamic leptin receptor (LepR) mRNA levels, which occurred in control animals when exposed to the HF diet, probably reflecting their higher resistance to obesity development. In addition, mRNA levels of suppressor of cytokine signalling 3 (SOCS3), a leptin-inducible inhibitor of leptin signalling and a potential mediator of leptin resistance in obesity(Reference Bjorbaek, Elmquist and Frantz16), were lower in leptin-treated animals than in the controls, when fed either a normal-fat (NF) or a HF diet(Reference Pico, Oliver and Sanchez11). Together, these results led to the postulate that leptin treatment during lactation may affect early programming mechanisms in the leptin and insulin signalling systems, resulting in adaptive changes in the control of food intake, which help to better regulate energy balance in adulthood(Reference Palou and Pico5), although specific mechanisms involved were not known.
DNA methylation is one of several epigenetic mechanisms that play a regulatory role in metabolic programming during the perinatal period(Reference McKay, Williams and Mathers17, Reference Mathers18). Increased methylation within the promoter regions correlates with transcriptional silencing, and the methylation status of CpG islands is believed to regulate gene transcription through the inhibition of transcription factors binding either directly or via altered histone acetylation(Reference Geiman and Robertson19). A range of environmental factors (including dietary components) have been shown to influence methylation patterns of CpG islands within the promoter regions of genes and thus influence gene expression (see Mathers & Ford(Reference Mathers, Ford, Choi and Friso20) for a review). This has led to the hypothesis that epigenetics may provide a plausible mechanistic link between the environment (particularly food/nutritional exposure) and alterations in gene expression that might lead to healthy or unhealthy phenotypes, and thus enable phenotypic plasticity in the context of a fixed genotype(Reference Jirtle and Skinner21). However, with a few exceptions, the genes influenced by these exposures remain largely unknown.
POMC expression has been reported to be strongly influenced by promoter methylation in different tissues(Reference Newell-Price, King and Clark22). The promoter of this gene is embedded within a defined CpG island, and much of this CpG island is methylated in normal non-expressing tissues, while it is unmethylated in expressing tissues, tumours and POMC-expressing cell lines(Reference Newell-Price, King and Clark22). In addition, Pomc promoter methylation in the hypothalamus has been shown to be sensitive to nutritional conditions during early life. In particular, neonatal overfeeding in rats, obtained by reducing the number of pups per litter, is associated with hypermethylation of CpG dinucleotides within the Pomc promoter(Reference Plagemann, Harder and Brunn23). There is no published information on the influence of the methylation status within the promoter regions of the Lepr and Socs3 genes.
Given that changes in methylation could be responsible for changes in gene expression and hence in the phenotype, we aimed to test the hypothesis that the pattern of methylation in the promoter regions of hypothalamic genes involved in appetite regulation and body weight control, namely Pomc, Lepr and Socs3, would be influenced by leptin treatment during lactation, particularly when exposed to a HF diet from weaning, and would be correlated inversely with expression of the same genes. Such epigenetic programming could explain potentially the better control of food intake observed following leptin treatment.
Methods
Animals and experimental design
The study was performed using thirty pups from six different dams, following the same protocol during lactation as described previously for the same cohort of animals(Reference Pico, Oliver and Sanchez11). Briefly, 3-month-old virgin female Wistar rats were mated with male rats (Charles River Laboratories, Barcelona, Spain). After mating, each female was placed in an individual cage with free access to water and food. Rats were kept in a room with controlled temperature (22°C) and a 12 h light–12 h dark cycle (lights on from 08.00 to 20.00 hours). At day 1 after delivery, excess pups in each litter were removed to keep ten pups per dam, and they were randomly assigned into two groups: control group and leptin-treated group. From day 1 to 20 of lactation, and during the first 2 h of the beginning of the light cycle, 20 μl of the vehicle (water, control group) or a solution of recombinant murine leptin (PeproTech, London, UK) dissolved in water (leptin-treated group) were given orally every day to the pups using a pipette. The amount of leptin given to the animals was calculated as five times the average amount of the daily leptin intake from the mother's milk(Reference Sanchez, Oliver and Miralles24). The exact daily doses for the consecutive 20 d were 1·0, 2·0, 3·0, 4·0, 5·0, 6·3, 7·5, 8·8, 10·0, 11·3, 15·6, 17·2, 18·8, 20·3, 21·9, 23·5, 25·0, 26·6, 39·4 and 43·8 ng. We considered these doses of leptin to be close to physiological levels of intake, taking into account the range of variation of milk leptin levels in dams(Reference Sanchez, Oliver and Miralles24). On day 21, after weaning, both control and leptin-treated male rats were single-caged and were divided into two groups: NF group – fed on a standard chow diet (15·89 kJ/g (3·8 kcal/g)) with 10 % energy from fat; and HF group – fed on a chow diet (19·66 kJ/g (4·7 kcal/g)) with 45 % energy from fat (Research Diets, Inc., New Brunswick, NJ, USA). The NF diet contained 5·5 % of energy from soyabean oil and 4·5 % from lard; the HF diet contained 5·5 % of energy from soyabean oil and 39·5 % from lard. Possible differences between litters were solved by ensuring the presence of pups from the same litter in each of the four experimental groups. Body weights and food intake were recorded three times/week from weaning until the age of 6 months.
At the age of 6 months, animals were killed by decapitation under fed conditions, during the first 2 h of the beginning of the light cycle, and the hypothalamus was removed rapidly. The whole hypothalamus was harvested using the following landmarks: frontal edge of the optical chiasm, lateral sulci, caudal edge of the mammary bodies, and a depth of 2 mm. These samples were immediately frozen in liquid N2 and stored at − 70°C. The animal protocol followed in the present study was reviewed and approved by the Bioethical Committee of University of the Balearic Islands and the guidelines for the use and care of laboratory animals of this University were followed.
DNA extraction and sodium bisulphite pyrosequencing
Total DNA was extracted from the hypothalamus by Tripure Reagent (Roche Diagnostic Gmbh, Mannheim, Germany), according to the manufacturer's instructions.
The promoter region of Pomc was obtained from the literature(Reference Jeannotte, Trifiro and Plante25) and those of Lepr and Socs3 were estimated using the Genomatix Gene2Promoter (Genomatix Software GmbH, Ann Arbor, MI, USA) software package. Nucleotide sequences of the rat Pomc, Lepr and Socs3 gene promoters were obtained from GenBank (National Center for Biotechnology Information, Bethesda, MD, USA). Putative transcription factor binding sites were predicted using Genomatix Matinspector (Genomatix Software GmbH). The location of each CpG Island was mapped by CpGIE (European Bioinformatics Institute, Hinxton, Cambridgeshire, UK), a java software program developed for CpG island identification.
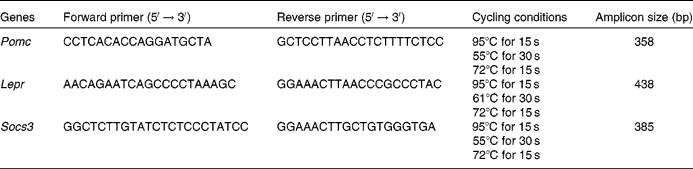
DNA methylation was measured by the bisulphite conversion method followed by pyrosequencing. Sodium bisulphite modification was performed using the EZ DNA Methylation-Gold Kit™ (Zymo Research, Orange, CA, USA), according to the manufacturer's instructions. Bisulphite-treated DNA was eluted in 10 μl of elution buffer. Modified DNA was amplified using PCR primers designed using PSQ Assay Design software (Biotage, Uppsala, Sweden; Table 1). Amplicons were generated in a 25 μl reaction volume containing 10 pmol of each PCR primer, 12·5 μl of Qiagen Hotstar Taq (QIAGEN GmbH, Hilden, Germany) and 10·5 μl water. Amplification was carried out in a G-storm thermocycler (GRI Limited, Wallingford, Oxon, UK) using the following protocol: 95°C for 15 min, then forty cycles of 95°C for 15 s, annealing temperature for 30 s, 72°C for 15 s, followed by 72°C for 5 min. Annealing temperatures were 47, 50 and 45°C for Pomc, Lepr and Socs3, respectively. All samples were analysed in duplicate. Based on the concentration of the PCR product, 5–10 μl PCR product were used for each pyrosequencing reaction. Pyrosequencing methylation analysis was carried out using the Pyro Q-CpG system (PyroMark; Biotage, Uppsala, Sweden), according to the manufacturer's protocol. In brief, the PCR product was bound to streptavidin-coated sepharose beads (GE Healthcare Bio-Sciences AB, Uppsala, Sweden), then washed in 70 % ethanol for 5 s, denatured in Denature buffer for 5 s and washed with washing buffer for 5 s using the PyroMark Q96 Vacuum Prep Workstation (Biotage). Then, a 0·5 μm sequencing primer was annealed to the purified single-stranded PCR product and pyrosequencing was carried out using the PyroMark Gold Reagents (Biotage). CpG site quantification was performed with the methylation software PyroQ-CpG™ (QIAGEN GmbH, Hilden, Germany). Unmethylated and in vitro methylated placental genomic DNA were analysed simultaneously as negative and positive controls, respectively.
Table 1 PCR primers and sequencing primers used in pyrosequencing assays for each gene

Pomc, pro-opiomelanocortin; F, forward; R, reverse; Lepr, leptin receptor; Socs3, suppressor of cytokine signalling 3.
Validity and sensitivity of the pyrosequencing assays
Calibration curves to assess the validity and sensitivity of the pyrosequencing assays were performed as described previously by White et al. (Reference White, Durston and Harvey26). Using placental DNA, an initial amplification step with control primers to unmethylate the sequence flanking the region of interest was carried out for each gene. The primers and the cycling conditions are described in Table 2. Then, half of the DNA product was artificially methylated with SssI (CpG) methylase (New England Biolabs, Hitchin, UK) and S-adenosylmethionine, according to the enzyme manufacturer's instructions. Following bisulphite modification, a dilution series of artificially methylated DNA in unmethylated DNA was used and mixed before PCR to generate samples with 0–100 % methylation. The samples were PCR-amplified and subjected to pyrosequencing. To ensure that the pyrosequencing assays provided unbiased quantification, we mixed PCR-amplified products generated from both methylated and unmethylated controls before pyrosequencing to generate samples with 0–100 % methylation.
Table 2 PCR control primers for validation assays
Pomc, pro-opiomelanocortin; Lepr, leptin receptor; Socs3, suppressor of cytokine signalling 3.
Statistical analysis
Data are expressed as means with their standard errors. To assess statistical significance, two-way ANOVA with the fixed factors of leptin treatment and diet was performed. Single comparisons between groups were assessed by Student's t test. A multiple correlation analysis of the studied parameters was also carried out and the Pearson's correlation index is given. The analyses were performed with SPSS for Windows (SPSS, Chicago, IL, USA). Threshold of significance was defined at P < 0·05.
Results
Body weight, food intake and circulating parameters
As described previously in the same cohort of animals(Reference Pico, Oliver and Sanchez11), animals given leptin-treatment during lactation had lower body weight in adulthood compared with untreated controls (Table 3). This protective effect was observed with both NF and HF diets. There were no detectable effects of leptin treatment on body weight during the lactation period (data not shown).
Table 3 Body weight and serum levels of glucose, insulin and leptin at the age of 6 months and cumulative food intake from day 21 to 6 months of life of male Wistar rats that received a daily oral dose of leptin or the vehicle during lactation, and were fed after weaning with a normal-fat (NF) or a high-fat (HF) diet*
(Mean values with their standard errors, n 6–8)

D, effect of the type of diet by two-way ANOVA (P < 0·05); L, effect of leptin treatment by two-way ANOVA (P < 0·05).
* Serum glucose was measured enzymatically using a kit, and leptin and insulin using an ELISA kit.
The effect of leptin treatment on body weight can be explained, at least in part, by lower food intake (Table 3). As described previously, cumulative food intake from weaning (at 21 d of age) to 6 months of age was significantly lower in leptin-treated animals v. their controls (P < 0·05, two-way ANOVA). Both control and leptin-treated animals consumed more energy when offered the HF than the NF diet (P < 0·05, two-way ANOVA).
Circulating glucose and insulin concentrations in adulthood were not significantly different between control and leptin-treated animals (Table 3), as described previously(Reference Pico, Oliver and Sanchez11). Serum insulin concentration was higher in control and leptin-treated animals under the HF diet, compared with NF diet-fed animals (P < 0·05, two-way ANOVA). Circulating leptin concentration was slightly reduced in leptin-treated rats v. their controls, both under the NF and HF diets, although differences did not reach statistical significance (Table 3).
Validation of the DNA methylation assays
CpG dinucleotides within the Pomc, Lepr and Socs3 promoters were mapped. For each assay, there was a linear decrease in the measured level of CpG methylation with dilution of the methylated DNA with unmethylated DNA, indicating that the assay provides accurate determination of CpG island methylation over several orders of magnitude, and also demonstrating an absence of any bias of unmethylated over methylated alleles introduced by PCR amplification.
DNA methylation pattern of CpG sites within the hypothalamic Pomc promoter
As described, the Pomc promoter spans over 404 bp and includes twenty-one CpG dinucleotides(Reference Newell-Price, King and Clark22). Here, we analysed six CpG sites located from − 271 to − 165 bp upstream from the transcription starting site ( − 238, − 224, − 216, − 202, − 192 and − 166). A total of twenty-five transcription factor binding sites were detected within the region that we analysed using Genomatix Matinspector (Genomatix Software GmbH), although only three of these sites had the potential to be affected directly by DNA methylation, namely ZFP161-, PAX5- and MYB-binding sites at CpG sites 2, 3 and 4 (Fig. 1(a)). Moreover, this region, and particularly CpG site 6 (which is closest to the transcriptional start site), corresponds to the region upstream of a Sp1-binding site, which has been shown to influence Sp1-induced gene transcription(Reference Zhu, Srinivasan and Dai27). In NF control animals, the percentage of methylation of the different CpG sites analysed varied between 24·6 (sem 1·9) and 34·2 (sem 2·3), with a mean of 29·2 (sem 1·2) (Fig. 1(b)).

Fig. 1 (a) Nucleotide sequence of the CpG island in the pro-opiomelanocortin (Pomc) promoter region showing individual CpG dinucleotides and putative transcription factor binding sites. (b) Methylation of individual CpG dinucleotides in the Pomc promoter in the hypothalamus of the normal-fat diet-fed controls. Values are means, with their standard errors represented by vertical bars, (n 5). NK6, NK6 homeobox transcription factors; POU1, GHF-1 (growth hormone factor 1) pituitary-specific pou domain transcription factor; PDX1, pancreatic and intestinal homeodomain transcription factor; LHX, lim homeodomain factors; BRN, Brn POU domain factors; CART, cartilage homeoprotein 1; NKX, NKX homeodomain factors; DLX, distal-less homeodomain transcription factors; BSX, brain-specific homeobox; HOX, paralogue hox genes 1–8 from the four hox clusters A, B, C, 6 I D; NK1, NK1 homeobox transcription factors; PAX, PAX (paired box gene) homeodomain-binding sites; OCT1, octamer-binding protein; ARID, AT-rich interactive domain factor; HBOX, homeobox transcription factors; TBP, vertebrate TATA-binding protein factor; BSN1, basonuclein rDNA transcription factor (PolI); ZFP161, zinc finger protein 161; PAX5, PAX-5 B-cell-specific activator protein; MYB, cellular and viral myb-like transcriptional regulators; STAT, signal transducer and activator of transcription; RP58, RP58 (ZFP238) zinc finger protein; NeuroD, NeuroD, Beta2, HLH (helix-loop-helix) domain.
The effect of feeding the HF diet on Pomc promoter methylation differed between control and leptin-treated animals. Specifically, across all six CpG sites assessed, methylation tended to increase in control animals fed the HF diet, but tended to decrease in leptin-treated animals under these dietary conditions (P = 0·086, two-way ANOVA, for the mean values of methylation across all six CpG sites). This effect was most significant at CpG site 6 (located at − 166 bp upstream from the transcription initiation site) (P = 0·057, interaction between leptin treatment and diet, two-way ANOVA; Fig. 2(a)). Given this evidence for a potential interaction between diet and leptin treatment (albeit that the P value for this interaction (P = 0·057) did not meet the conventional cut-off for significance (P < 0·05)), we explored the differences between individual treatment groups using Student's t test. For this CpG site, under the NF diet, methylation was higher in leptin-treated animals compared with controls (P = 0·034, Student's t test). In contrast, with HF diet feeding, there was a significant decrease in methylation in leptin-treated animals (P = 0·035, Student's t test), while methylation tended to increase in control animals. However, until confirmed by independent studies, these findings should be treated with caution.

Fig. 2 (a) Percentage of methylation of the six CpG sites studied for the pro-opiomelanocortin (Pomc) gene of male Wistar rats that received a daily oral dose of leptin or the vehicle during lactation, and were fed after weaning with a normal-fat (NF) or a high-fat (HF) diet. Values are means, with their standard errors represented by vertical bars (n 5–8). L × D, interaction between leptin treatment and the type of diet (P < 0·05, two-way ANOVA). *NF v. HF diet; †control v. leptin (Student's t test). □, NF control; ![]() , HF control;
, HF control; ![]() , NF leptin;
, NF leptin; ![]() , HF leptin. (b) Correlation between hypothalamic POMC mRNA expression levels (expressed relative to the value of control animals under the NF diet, which was set to 100 %) and percentage of methylation of CpG site 6 (located at − 166 upstream from the transcription starting site), considering the four groups of animals described above. Simple correlations were assessed by Pearson's correlation coefficients. The r and P values for correlations are − 0·318 and 0·075, respectively.
, HF leptin. (b) Correlation between hypothalamic POMC mRNA expression levels (expressed relative to the value of control animals under the NF diet, which was set to 100 %) and percentage of methylation of CpG site 6 (located at − 166 upstream from the transcription starting site), considering the four groups of animals described above. Simple correlations were assessed by Pearson's correlation coefficients. The r and P values for correlations are − 0·318 and 0·075, respectively.
We then used correlation analysis to investigate relationships between methylation at individual CpG sites within the Pomc promoter and Pomc gene expression, as determined previously(Reference Pico, Oliver and Sanchez11). No significant correlations were found between promoter methylation and POMC mRNA levels, but a weak (non-significant) negative correlation was found for all CpG sites investigated. The most significant relationship was observed between gene expression and methylation at CpG site 6 (P = 0·075; Fig. 2(b)).
DNA methylation pattern of CpG sites within the hypothalamic Lepr promoter
For the Lepr promoter, a sequence located from − 10 170 to − 9894 bp upstream from the transcription starting site was analysed, containing a total of nineteen CpG dinucleotides ( − 9914, − 9961, − 9980, − 9993, − 9997, − 10 004, − 10 015, − 10 040, − 10 042, − 10 091, − 10 120, − 10 137, − 10 147, − 10 150, − 10 152, − 10 156, − 10 158, − 10 164 and − 10 166) (Fig. 3(a)), and thirty-one putative transcription binding sites were detected using the Genomatix Matinspector (Genomatix Software GmbH). In NF control rats, the percentage of methylation of the different CpG sites analysed varied between 34·4 (sem 8·9) and 47·3 (sem 12·6), with a mean of 41·5 (sem 10·7) (Fig. 3(b)).

Fig. 3 (a) Nucleotide sequence of the CpG island in the leptin receptor (Lepr) promoter region showing individual CpG dinucleotides and putative transcription factor binding sites. (b) Methylation of individual CpG dinucleotides in the Lepr promoter in the hypothalamus of the normal-fat diet-fed controls. Values are means, with their standard errors represented by vertical bars (n 5). PAX9, PAX-9 binding sites; MTEN, core promoter motif ten elements; HES, vertebrate homologues of enhancer of split complex; NRF1, nuclear respiratory factor 1; E2F, E2F-myc activator/cell cycle regulator; ZBP, zinc-binding protein factors; RBP, retinoblastoma-binding proteins with demethylase activity; NR2, nuclear receptor subfamily 2 factors; HAND, twist subfamily of class B bHLH (helix-loop-helix) transcription factors; GRE, glucocorticoid responsive and related elements; NF1, nuclear factor 1; SP1, GC-box factors SP1/GC; XCPE, activator-, mediator- and TBP-dependent core promoter element for RNA polymerase II transcription from TATA-less promoters; SF1, vertebrate steroidogenic factor; NRS, neuron-restrictive silencer factor; CTC, CTCF (CCCTC-binding factor) and BORIS (CCCTC-binding factor (zinc finger protein)-like) gene family, transcriptional regulators with eleven highly conserved zinc finger domains; PAR, PAR/bZIP (basic-leucine zipper) family; XBOX, X-box-binding factors; NFAT, nuclear factor of activated T-cells; AP2, activator protein 2; INS, insulinoma-associated factors; HOM, homeodomain transcription factors; LHX, lim homeodomain factors.
No significant (P>0·05) effects of leptin treatment during lactation or of the type of diet given post-weaning on the methylation status of any CpG sites within the Lepr promoter were detected (two-way ANOVA, data not shown).
DNA methylation pattern of CpG sites within the hypothalamic Socs3 promoter
For the Socs3 promoter, thirteen CpG dinucleotides located from − 316 to − 125 bp upstream from the transcription starting site were analysed ( − 137, − 143, − 158, − 165, − 187, − 210, − 219, − 238, − 248, − 292, − 306, − 308 and − 310). In this sequence, seventeen possible transcription factor binding sites were predicted with the Genomatix Matinspector (Genomatix Software GmbH) (Fig. 4(a)). The percentage of methylation in the NF control rats varied between 43·5 (sem 6·6) and 69·0 (sem 9·4) throughout the different CpG dinucleotides analysed, with a mean of 58·0 (sem 8·2) (Fig. 4(b)).

Fig. 4 (a) Nucleotide sequence of the CpG island in the suppressor of cytokine signalling (Socs3) promoter region showing individual CpG dinucleotides and putative transcription factor binding sites. (b) Methylation of individual CpG dinucleotides in the Socs3 promoter in the hypothalamus of the NF diet-fed controls. Values are means (n 5), with their standard errors represented by vertical bars. PAX4/PAX6, PAX-4/PAX-6 paired domain-binding sites; EBOX, E-box-binding factors; ETS1, human and murine ETS1 factors (v-ets erythroblastosis virus E26 oncogene homolog 1 (avian)); INS, insulinoma-associated factors; HAM, human acute myelogenous leukaemia factors; XBB, X-box-binding factors; RXR, retinoid X receptor heterodimer-binding sites; MTEN, core promoter motif ten elements; PAX5, PAX-5 B-cell-specific activator protein; CTC, CTCF (CCCTC-binding factor) and BORIS (CCCTC-binding factor (zinc finger protein)-like) gene family, transcriptional regulators with eleven highly conserved zinc finger domains; ROR, v-ERB and RAR (retinoic acid receptor)-related orphan receptor α; NR2, nuclear receptor subfamily 2 factors; LEF, lymphoid enhancer-binding factor 1; KLF, Krueppel-like transcription factors; HIC1, Krueppel-like C2H2 zinc finger factors hypermethylated in cancer; AP2, activator protein 2; SPZ1, testis-specific bHLH-Zip transcription factors.
No significant (P>0·05) effects of leptin treatment during lactation or of the type of diet given post-weaning on the methylation status of any CpG sites within the Socs3 promoter were detected (two-way ANOVA, data not shown).
Discussion
It is becoming increasingly clear that environmental factors during perinatal life are important determinants of lifelong health. In this context, leptin, a natural component of breast milk(Reference Casabiell, Pineiro and Tome7, Reference Houseknecht, McGuire and Portocarrero8), that is not present in infant formula(Reference Resto, O'Connor and Leef28), has been identified recently as a specific milk component that could be responsible, at least in part, for the beneficial effects of breast-feeding compared with formula feeding, against the development of obesity and other features of the metabolic syndrome in adulthood(Reference Palou and Pico5, Reference Pico, Oliver and Sanchez11). The mechanisms responsible for the protective effects of leptin during lactation on lifelong health are not known, although several hypotheses are emerging.
Leptin seems to play a role during a critical developmental window to ensure the normal development of hypothalamic pathways in the arcuate nucleus, which are important because they convey leptin signals to brain regions regulating body weight and, therefore, regulate the impact of leptin on energy homeostasis throughout life(Reference Bouret, Draper and Simerly29). Leptin-deficient (Lepob/Lepob) mice present an altered hypothalamic development characterised by a dramatic decrease in neuronal fibre density in hypothalamic structures involved in leptin signalling(Reference Bouret, Draper and Simerly29).
In addition, leptin treatment during lactation has lasting effects on the expression of hypothalamic factors involved in the control of food intake and regulated at the central level by leptin, particularly POMC, LepR and SOCS3, when animals are exposed to a HF diet(Reference Pico, Oliver and Sanchez11). This pattern of expression may contribute to the apparently improved capacity to regulate food intake, even when exposed to HF diets, thus helping to protect animals against excess weight gain in adulthood. In the first study of its kind, we have attempted to test the hypothesis that the observed changes in the expression of three hypothalamic genes, namely Pomc, Socs3 and Lepr, are associated with changes in the promoter methylation of these genes. Since DNA methylation marks are known to be plastic in response to dietary and other environmental exposures and changes in promoter methylation may result in altered gene expression, such epigenetic mechanisms may mediate the lifelong effects of early exposures on health outcomes such as obesity (see Mathers(Reference Mathers, Dube, Bechara, Dagher, Drewnowski, LeBel, James and Yada30) for a review). Here, we show that leptin and dietary treatment induce changes in the methylation of CpG sites in the Pomc promoter. Although the available evidence is modest, these changes in Pomc promoter methylation explain, in part, the changes observed in POMC mRNA expression levels in leptin-treated animals, particularly when these animals were exposed to the HF diet (see Fig. 2 showing a weak negative correlation between methylation status and mRNA expression). The effects of leptin and dietary treatment on promoter methylation were relatively modest but they provide proof of principle that such exposures may be ‘recorded’ as altered patterns of DNA methylation in brain regions such as the hypothalamus, which are critical for the regulation of energy homeostasis. There is good evidence that increased promoter methylation and reduced gene expression are linked causally for a limited number of tumour suppressor genes and there is growing evidence of inverse associations for a number of age-related genes(Reference Ronn, Poulsen and Hansson31, Reference Maegawa, Hinkal and Kim32). However, as yet, there is limited understanding of which CpG sites within the promoter regions of genes are sensitive to the effects of environmental exposures and the extent to which methylation at these CpG sites regulates gene expression. In the present initial study, we took a pragmatic approach and tested a relatively small panel of CpG sites. Future studies will require more systematic analysis of the regulatory elements within genes.
Similarly, although the present study found no effects of leptin or dietary fat on the methylation status of Lepr and Socs3, it is possible that the particular sites studied within the promoter region of these genes were not the most informative and we cannot exclude the possibility that expression of these genes was regulated by methylation changes in other regions of the promoters of these genes. Nevertheless, to the best of our knowledge, no data exist regarding the association between changes in the methylation status of these genes and changes in their mRNA expression levels as a result of perinatal conditions, although this is a relatively unexplored field.
POMC is the precursor of α-melanocyte-stimulating hormone, which is a potent inhibitor of food intake and is considered to be the predominant pathway regulating food intake in adults(Reference Bell, Bhatnagar and Akana33). Hypothalamic POMC expression is increased in the presence of elevated levels of the anorexigenic hormones leptin and insulin(Reference Schwartz, Woods and Porte14). The promoter region studied here, and particularly the region containing the − 166 CpG site, is located just upstream of a Sp1-binding site. Sp1 is a major activator of Pomc transcription(Reference Therrien and Drouin34), and activation of this Sp1-binding site in the Pomc promoter has been described to be essential for the mediation of leptin effects on POMC expression(Reference Yang, Lim and Li35). In addition, the expression of Sp1 is physiologically stimulated by insulin(Reference Pan, Solomon and Borromeo36). Therefore, changes in the methylation status in the Pomc promoter region affecting Sp1 binding may be essential for the mediation of leptin and insulin effects on POMC expression, and therefore could contribute to the perinatal programming of sensibility to these hormones. Of note, methylation upstream of the Sp1-binding site, as distinct from methylation within the consensus Sp1-binding site itself, has been shown to inhibit Sp1 binding(Reference Zhu, Srinivasan and Dai27). Therefore, the hypomethylation of CpG dinucleotides within the Pomc promoter in a region upstream of the Sp1-binding site found in leptin-treated animals under HF diet feeding conditions is in accordance with their higher expression levels of POMC in a situation of increased levels of the peripheral anorexigenic signals insulin and leptin (although for the latter, the increase did not reach statistical significance). Such increased POMC expression was not found in control animals. Thus, although we have not determined directly whether changes in the methylation status of this promoter region directly affect Sp1 binding and/or POMC expression, the present results suggest that changes in promoter methylation may be one of the mechanisms that affects the expression of this gene. This may explain an improved sensitivity to the insulin and leptin action that may be responsible for the relative resistance to the hyperphagia and the accumulation of excess fat associated with HF diet exposure. Other critical factors, including circulating levels of leptin and insulin and the abundance of their receptors, and other epigenetic mechanisms, e.g. changes in microRNA or in post-translational modifications of histones, may contribute to altered expression of POMC at the mRNA expression level in response to perinatal leptin treatment.
In addition to the possible involvement of Sp1, other CpG sites within the Pomc promoter, which we analysed, may be target binding sites for the transcription factors ZPF161, PAX5 and MYB. However, the putative involvement of these transcription factors in the regulation of POMC expression and whether their binding may be affected by the methylation status have not been studied, although these do not seem evident from the present studies.
Changes in Pomc promoter methylation in the hypothalamus as a consequence of environmental conditions during early life have also been described recently in a model of overfeeding during lactation, obtained by rearing rats in small litters(Reference Plagemann, Harder and Brunn23). These conditions have been reported to lead to rapid early weight gain, resulting in a metabolic syndrome phenotype, including obesity, hyperleptinaemia and hyperglycaemia(Reference Plagemann, Harder and Brunn23, Reference Plagemann, Harder and Rake37). In these animals, the hypothalamic Pomc promoter showed hypermethylation of CpG dinucleotides upstream of the Sp1-binding sequence ( − 156 to − 152), and this was associated with a lack of Pomc up-regulation, despite hyperleptinaemia and hyperinsulinaemia(Reference Plagemann, Harder and Brunn23). Moreover, changes in Pomc promoter methylation have also been described in a model of intra-uterine growth retardation due to protein restriction(Reference Coupe, Amarger and Grit38). Rats that were protein restricted during gestation showed a higher methylation level than their controls in the CpG site at position − 224 of the Pomc gene(Reference Coupe, Amarger and Grit38). In addition, animals that were restricted both during gestation and lactation were less methylated than controls for the CpG sites located at − 216 and − 164(Reference Coupe, Amarger and Grit38). Altogether, these results suggest that both pre- and postnatal periods may influence the methylation level at some CpG sites of the Pomc promoter in the hypothalamus, as well as emphasise the potential importance of nutritionally induced alterations in hypothalamic Pomc methylation for body-weight regulation and obesity programming.
It must be noted that changes in Pomc promoter methylation as an effect of leptin treatment were manifested when animals were under chronic HF diet conditions, whereas no differences were found under NF diet conditions. The HF diet was offered to animals after weaning and continued during the whole period studied. This means that these changes in Pomc promoter methylation occurred in the post-weaning period, and not during the suckling period, and suggests that patterns of Pomc promoter methylation may remain plastic into adulthood. However, it is also apparent that leptin treatment during lactation may alter susceptibility to later epigenetic changes. With the present study design, we cannot specify when these changes occurred, i.e. which period of life is critical for these particular epigenetic changes. It has been proposed that epigenome stability is decreased during the pubertal period(Reference Jirtle and Skinner21), thus interventions applied during this part of the life course may have the potential to modify phenotypes induced in early life and thereby change lifelong risk of disease. Nevertheless, the brain appears to retain considerable plasticity with respect to DNA methylation marks, and changes in DNA methylation patterns in the brain throughout life have been reported(Reference Siegmund, Connor and Campan39). On the other hand, there is an apparent contradiction regarding both mRNA levels and DNA methylation of Pomc in NF leptin-treated rats. In fact, the lower body weight and food intake of NF leptin-treated animals compared with their controls cannot be explained by the lower mRNA expression levels of POMC, suggesting that other mechanisms affected by leptin supplementation during lactation may counteract the lowered POMC mRNA expression. Further studies are needed to elucidate these observations.
In conclusion, results presented here show that leptin treatment during lactation promotes epigenetic modification in the Pomc promoter with lasting effects on food intake and body weight, particularly when these animals are exposed to a HF diet. These data give some insight into the possible mechanisms affecting gene expression in the central nervous system in animals that underwent oral leptin treatment during the suckling period. It remains to be determined how leptin supplementation during the suckling period is able to induce these epigenetic effects on DNA methylation, as well as to perform a more systematic examination of CpG sites in the regulatory regions of these genes.
Acknowledgements
The authors declare no conflict of interest. We gratefully acknowledge the Spanish Government (grant no. AGL2009-11277), the BBSRC (grant no. BB/G007993/1) and the European Research Network of Excellence NuGO (The European Nutrigenomics Organization, European Union contract no. FP6-506360). The CIBER de Fisiopatología de la Obesidad y Nutrición is an initiative of the ISCIII. M. P. carried out the methylation analyses and participated in the analyses, interpretation and discussion of the results. C. P. participated in the experimental design of the study and in its coordination, carried out the discussion of the results and wrote the first version of the manuscript. J. A. M. contributed to the design and performance of methylation analyses and explained these procedures to M. P. J. S. carried out the animal treatment, follow-up and sample collection and performed the analysis of blood parameters. T. P. participated in sample collection and performed the gene expression analyses. J. C. M. participated in the design and coordination of the methylation studies and wrote the final version of the manuscript. A. P. conceived and co-directed the study, participated in its design and coordination and revised the final version of the manuscript. All authors read and approved the final manuscript.







