


INTRODUCTION
Tuberculosis (TB) is an infectious and chronic disease affecting a wide range of mammalian species. Within those species, swine are susceptible to infection with a variety of Mycobacteria, including Mycobacterium bovis, M. tuberculosis, and M. avium complex (MAC) [Reference Matlova1–Reference Arega, Conraths and Ameni3]. TB infection affects animal production because of its economic impact and its potential risk for zoonotic transmission [Reference Mijs4, Reference Agdestein5]. Because mycobacterial infection in pigs normally does not produce a clinical manifestation of disease, TB is usually only detected by the presence of macroscopic lesions during slaughterhouse inspection. MAC infection typically causes localized, proliferative lesions and usually affects the submandibular and mesenteric lymph nodes [Reference Matlova1, Reference Domingos, Amado and Botelho6, Reference Perez7]. On the other hand, M. bovis infection typically produces caseous and necrotic lesions in the lungs, spleen and lymph nodes in the thoracic and/or abdominal cavity [Reference Perez7]. In countries in which bovine TB (bTB) is yet to be eradicated, disease prevalence in pigs is related to that found in cattle and is usually due to M. bovis [Reference Serraino8–Reference Muwonge10]. Conversely, in regions free from bTB, the disease in swine is more frequently associated with MAC [Reference Alvarez11, Reference Lara12].
MAC includes M. intracellulare, M. avium subsp. paratuberculosis (MAP), M. avium subsp. silvaticum (MAS), M. avium subsp. avium (MAA) and M. avium subsp. hominissuis (MAH). In particular, MAA and MAH are the M. avium subspecies that are most frequently associated with TB in pigs [Reference Alvarez11, Reference Lara12].
Before the use of molecular techniques, MAA was believed to be responsible for most of the MAC cases of infection in pigs. However, MAH infection in pigs and humans is more frequent than originally suspected [Reference Matlova1, Reference Agdestein5].
The molecular characterization of Mycobacteria is conducted using alternative polymerase chain reaction (PCR) target sequences. For instance, M. bovis is typically identified using the insertion sequence IS6110, followed by spoligotyping for between- and within-species differentiation [Reference Hermans13, Reference Kamerbeek14]. Regarding MAC, the insertion sequence IS1245 is characteristic of M. avium and is useful to discriminate between M. avium and M. intracellulare [Reference Guerrero15], whereas IS901 is present in MAA and MAS but is absent in MAH [Reference Mijs4, Reference Kunze16]. The molecular typing reference method for epidemiological links of M. avium is the restriction fragment length polymorphism (RFLP) with IS1245, IS1311 and IS900 as probes [Reference Domingos, Amado and Botelho6, Reference Johansen17]. However, in recent years, the development of molecular typing methods based on PCR has substantially simplified the studies of the TB infection.
Multilocus variable number tandem repeat analysis (MLVA) is based on the identification of mycobacterial repetitive elements referred to as a mycobacterial interspersed repetitive unit, which results in a variable number of tandem repeats (MIRU-VNTR). Nowadays, MIRU-VNTR analysis is increasingly used for typing M. avium isolates [Reference Thibault18–Reference Pate22]. A panel of eight MIRU-VNTR markers has been identified as a suitable tool to discriminate isolates and to study the genetic variability of the strains within the MAC.
M. bovis infection is more frequent than MAC infection in the pig population of Argentina [Reference de Kantor and Lesslie23–Reference Barandiaran25]. This may be the reason why MAC infection has been little characterized in the country.
The aim of this study was to characterize the epidemiology of MAC infection in a swine population of Argentina by means of molecular tools and spatial analysis techniques. Specifically, we (1) studied the frequency of TB cases caused by MAC infection, which was also stratified by type of Mycobacteria (i.e. MAA or MAH) and genotyped using MIRU-VNTR; (2) assessed if co-infection of MAC and M. bovis occurs in the field; and (3) described the relative frequency and spatial distribution of MAC genotypes. The results herein presented will be useful to characterize and quantify the impact of the disease and, ultimately, to promote disease control measures in the most important porcine production regions of Argentina.
METHODS
Samples
Pig carcasses were bromatologically inspected at slaughterhouses. The selection criteria of samples consisted of presence of macroscopic granulomatous lesions caseo-necrotic and/or proliferative. The head and mesenteric lymph nodes as well as the liver and lungs of the pigs were routinely examined; gross lesions in other locations were also recorded. About 10 g of the lymph nodes with gross pathological lesions were placed in a sterile bag and transported to the laboratory in a refrigerated container. The samples were stored at −20 °C for up 2 months until bacteriological processing.
Isolates
TB-like lesions (n = 200) were collected from May 2007 to November 2009 at three slaughterhouses located in Buenos Aires province, Argentina. The studied pigs came from herds located in three provinces (Buenos Aires, Santa Fe, Cordoba) of the Pampa region of Argentina, which accounts for most of the country's swine production. The samples were homogenized and decontaminated using Petroff's method [Reference Jorge26] with 4% sodium hydroxide followed by seeding for 8 weeks at 37 °C in Löwenstein–Jensen (LJ) and Stonebrink (ST) media to stimulate the developing of MAC and M. bovis, respectively. The cultures were examined weekly. A bacteriological typing of the isolates was performed based on the growing time, culture media, colony morphology, and the presence of acid-fast bacilli (AFB), which was detected through Ziehl-Neelsen staining [Reference Jorge26].
Molecular typing
The AFB-positive isolates were tested by PCR for species-specific identification of Mycobacteria. The PCR-targeted sequences were IS6110 for M. tuberculosis complex (CMTB) [Reference Hermans13]; IS1245 for M. avium [Reference Guerrero15], and IS901 for MAA and MAS, specifically [Reference Kunze16]. The IS6110-positive samples were subsequently subjected to spoligotyping to identify M. bovis (absence of 3, 9, 16 and 39–43 spacers) and for intraspecies differentiation [Reference Kamerbeek14].
Detection of co-infection
The positive samples were considered co-infected with M. bovis and MAC if the isolates developed in ST and/or LJ culture media and had a PCR-positive result for IS6110 and IS1245 along with spoligotypes characteristic of M. bovis. Although MAC is not a strict pathogen, which decreases the chances of environmental pollution, this possibility cannot be totally excluded. However, the isolation of bacteria was done from granulomatous lesions; which strengthen the idea that MAC is the infective agent. The criteria used to determine co-infection were confirmation by PCR of two species in suspensions from at least two colonies grown in culture media. We considered infection present when a single species of Mycobacteria was confirmed by PCR [Reference Matlova1, Reference Agdestein5].
Genotyping of M. avium strains
The MAC-positive isolates were typed using MIRU-VNTR. The scheme selected was based on eight MIRU-VNTR loci, namely, VNTR-292, MIRU-X3, VNTR-25, VNTR-47, VNTR-3, VNTR-7, VNTR-10, and VNTR-32 (simplified as 292, X3, 25, 47, 3, 7, 10, and 32, respectively). Primer sequences and PCR conditions were as described elsewhere [Reference Thibault18]. Molecular weights were calculated using BioNumerics software v. 3·5 (Applied Maths, Belgium) and the number of tandem repeats in each locus was assigned following a protocol described previously [Reference Radomski20]. Numerical profiles were classified according to INMV (INRA, Nouzilly, MIRU-VNTR) combinations [Reference Thibault18, Reference Radomski20].
Spatial distribution of MAC genotypes
Because the farm origin was unknown, the MAC-positive isolates were assumed to have originated from the centroid of the department in which the farm of origin was located. The multinomial model of the spatial scan statistic was used to identify if the samples belonging to a given INMV category were spatially clustered [Reference Jung, Kulldorff and Richard27]. The null hypothesis was that INMVs were randomly distributed in the study area, whereas the alternative hypothesis was that at least one of the INMVs was spatially clustered. The significance of clustering was tested using a simulation process and the multinomial model of the scan statistic was implemented using SaTScan software [Reference Kulldorff28].
RESULTS
A total of 216 mycobacterial strains were identified by PCR, of which 160 (74·07%), 16 (7·40%), and 20 (9·25%) were positive to M. bovis (IS6110), M. avium (IS1245), and both M. bovis and M. avium, respectively (Fig. 1). Mycobacterium isolates (n = 196) were obtained from most (98%) of the TB-like lesions assessed (n = 200). Of the 216 mycobacterial strains, 180 (83·33%) were M. bovis (IS6110) positive with typical spoligotypes, including 143 isolates described elsewhere [Reference Kulldorff28]; and 36 (16·66%) remaining strains were MAC (IS1245 positive). Of these remaining strains (n = 36), 30 (83·33%) were MAA (IS901 positive), whereas six (16·66%) were MAH (IS901 negative), following the selection criteria described by Eisenberg et al. [Reference Eisenberg29].
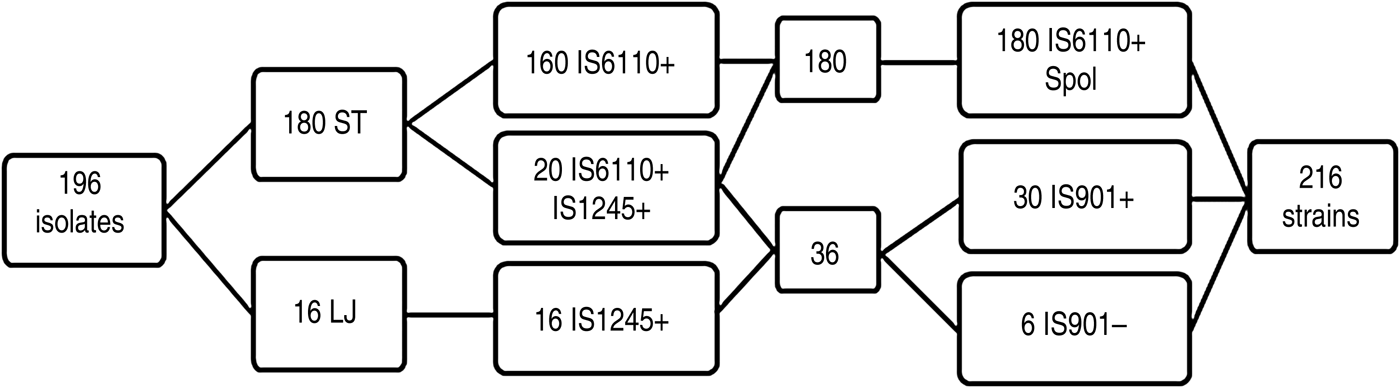
Fig. 1. Scheme of the results obtained by culture, polymerase chain reaction (PCR) and spoligotyping. ST, Stonebrink medium; LJ, Löwestein–Jensen medium; Spol, spoligotyping; IS, insertion sequence. Positive and negative PCR results are denoted by + or –, respectively.
Most (n = 180, 91·8%) of the isolates developed in ST, whereas the remaining 16 (8·2%) isolates developed in LJ. Furthermore, some (n = 20, 11·1%) of the isolates that developed in ST medium co-exist with two species (IS6110 PCR- and IS1245 PCR-positive). Conversely, all the isolates grown in LJ medium were IS6110 PCR-negative. Thus, M. bovis was detected only in ST medium, whereas M. avium was found in LJ (n = 16) and ST (n = 20) media. Multiple (n = 5) spoligotypes were detected in the 20 isolates where co-existance of M. bovis and MAC was detected (Table 1). All the six MAH isolates grew in ST medium and in co-existence with M. bovis.
Table 1. MIRU-VNTR types detected in the 29 M. avium subsp. avium and six M. avium subsp. hominissuis isolates. The co-existence of M. avium and M. bovis isolates is detailed. PCR and spoligotyping results are also shown
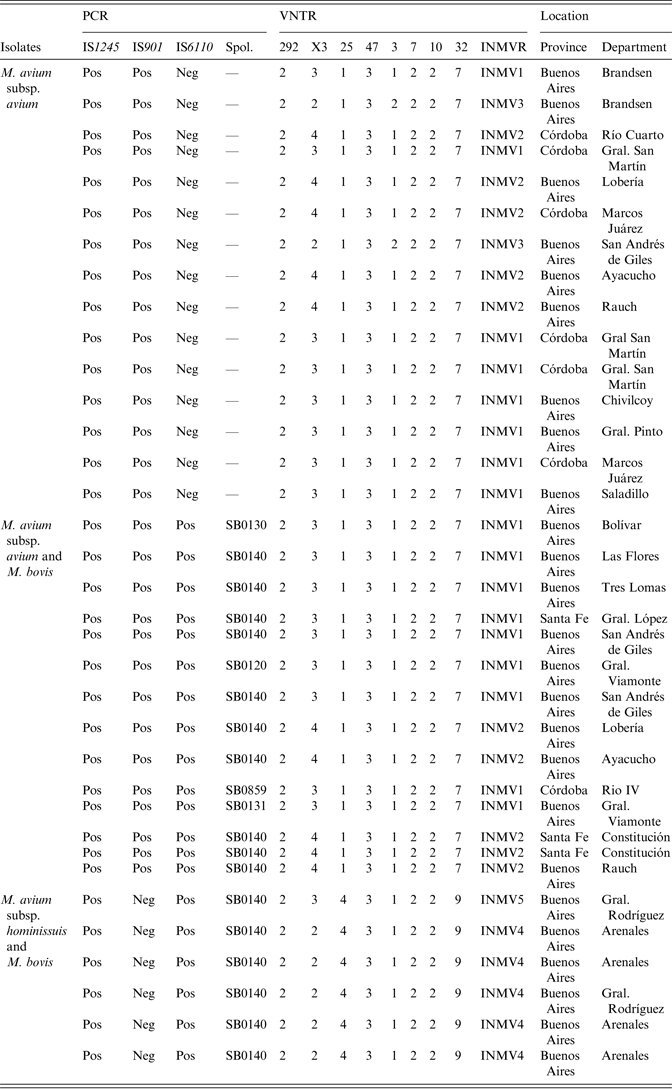
MIRU-VNTR, Mycobacterial interspersed repetitive unit, variable number of tandem repeats; PCR, polymerase chain reaction.
A MIRU-VNTR analysis revealed multiple patterns (n = 8) in 35 of the MAC-positive strains. Five different MIRU-VNTR patterns (referred to as INMVs) were observed (Table 1). The INMV1 and INMV2 patterns represented most isolates (48·5% and 28·5%, respectively), whereas INMV4, INMV3, and INMV5 grouped 14·2%, 5·7%, and 2·7% of the isolates, respectively. Most (n = 34, 97·14%) of the MAC isolates were grouped into four MIRU-VNTR clusters, whereas only one had a unique pattern (Table 1). Only one MAH strain did not belong to the INMV4 group. Furthermore, none of the patterns deduced for MAA matched those of MAH. In terms of the discriminatory power, locus X3 displayed the highest allelic diversity 0·63, followed by loci 25 and 32 with 0·29 for each (Table 2). Loci 292, 47, 7 and 10 lacked allelic diversity (Table 3).
Table 2. Allelic distribution of polymorphic MIRU-VNTR
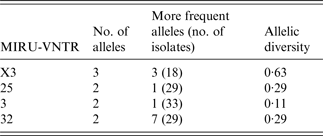
MIRU-VNTR, Mycobacterial interspersed repetitive unit, variable number of tandem repeats.
The discriminatory index of Hunter and Gaston was calculated using the formula of Simpson (http://insilico.ehu.es/mini_tools/discriminatory_power/).
Table 3. MIRU-VNTR allelic distribution of M. avium subsp. avium and M. avium subsp. hominissuis isolates
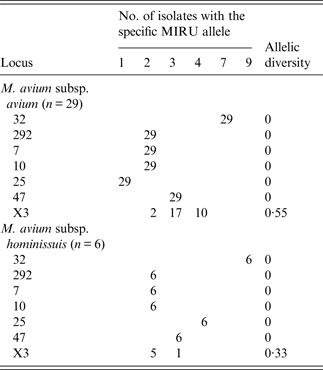
MIRU-VNTR, Mycobacterial interspersed repetitive unit, variable number of tandem repeats.
We detected a significant (P = 0·017) spatial cluster of INMVs, which included four MAH-positive samples located in General Arenales (Fig. 2). Interestingly, under the null hypothesis of homogeneous distribution of INMVs, the expected number of MAH-positive samples in this region, was 0·57 (observed-to-expected ratio = 7).

Fig. 2. Map of the central region of Argentina (Buenos Aires, Córdoba, Santa Fe, La Pampa, Entre Ríos provinces) showing the distribution of the isolates (ArcGIS, v. 10·0; ESRI, USA). Each department shows the number of isolates and the genotypes are denoted by greyscale. In General Arenales department, with four isolates, a significant spatial clustering of MAH was observed. INMV, INRA Nouzilly, MIRU-VNTR (numerical profile combinations).
DISCUSSION
In this study, we isolated strains of M. avium (n = 36) and M. bovis (n = 180) from granulomatous TB-like lesions collected from slaughtered domestic pigs. The predominant Mycobacteria in pigs depend on the epidemiological situation of TB in the country and the production characteristics of each region. An effective control of mycobacterial infections requires knowledge of the causative agent and its epidemiology as well as knowledge of interspecies transmission and biodiversity within the isolated strains [Reference Tirkkonen21].
Because bTB is endemic in Argentina, it was not surprising that M. bovis was the most frequently isolated bacterium in this study. However, a large proportion of MAC strains were also identified and the frequency of M. avium was larger (16·8%) than those reported in previous studies (6–7·5%) in Argentina [Reference de Kantor and Lesslie23, Reference Perez24]. This finding may be explained, at least in part, by the decrease in the prevalence of bTB in cattle over the last 43 years, from 6·7% in 1969 to 0·3% in 2012 [30]. Such an epidemiological feature may have resulted in a relative increase in the proportion of MAC isolates from swine granulomatous lesions, as has been observed in countries in which bTB has been eradicated [Reference Matlova1, Reference Agdestein5]. Specifically in Argentina, TB prevalence in pigs decreased from 8·4% in 1969 to 0·3% in 2012 [30]. Therefore, we hypothesize that the reduction of competition between M. bovis and MAC probably allowed MAC to occupy more space in the chain of infection.
MAA and MAH are the two species of highest epidemiological importance in swine. MAA is the causative agent of avian TB and in turn birds, together with environments contaminated with infected bird faeces, are the main source of infection for pigs [Reference Agdestein5, Reference Moravkova31]. On the other hand, MAH is the most prevalent opportunistic pathogen for pigs and humans, and has been isolated from many host species and environmental samples [Reference Matlova1, Reference Agdestein5, Reference Johansen17]. Furthermore, MAH has also acquired an increasing importance for public health in recent decades because of its ability to cause pulmonary disease, lymphadenitis in children and disseminated infections in immunocompromised patients [Reference Alvarez11]. In Argentina M. avium was reported as the major aetiological agent of clinically confirmed cases of non-tuberculous Mycobacteria in humans, but no differentiation of species and subspecies of MA has been assessed yet [Reference Imperiale32]. The use of molecular techniques had allowed the identification of a close genetic relationship between strains found in humans and swine, which suggest transmission between these two hosts or exposure to a common source of infection [Reference Johansen17, Reference Tirkkonen21, Reference Pate22]. Thus, pigs could be considered as an additional source of transmission of M. avium to humans, especially to immunosuppressed populations [Reference Johansen17, Reference Tirkkonen21, Reference Pate22]. MAH has previously been identified in two dogs (miniature Schnauzer) and one human in Argentina [Reference Martinez Vivot33, Reference Gioffré34].
As far as we know, this is the first report of MAH infection in pigs in Argentina. Most (66%) of the MAH isolates were clustered (P < 0·01) in one single department from Buenos Aires province, General Arenales, which suggests a limited geographical distribution of MAH in the country. Other M. avium strains (n = 30) were classified as MAA (IS901) positive. Some characteristics of swine production in Argentina may favour the transmission of these two species of Mycobacteria in swine. Outdoor production systems are relatively common and, in farms with limited or absent biosecurity measures in place, pigs may have contact with wild birds and/or wild boars, which increases the opportunity for disease transmission.
For MLVA analysis we used the set of primers designed on the basis of the MAP genomic sequence [Reference Thibault18] and detected multiple (n = 5) MLVA patterns. The most frequent pattern, referred to as INMV1, grouped 48·5% (17/35) of the isolates and included animals from the three sampled provinces (Buenos Aires, Córdoba, Santa Fe) (Table 1 and Fig. 2). The observed patterns were different compared to those found associated with poultry, cattle, and humans from Argentina [Reference Gioffré34]. However, the absence of such patterns may be due to the limited number of isolates that are available and analysed using this technique [Reference Gioffré34].
The highest polymorphism was observed in MAA isolates compared to MAH strains, in contrast to that observed elsewhere [Reference Radomski20, Reference Pate22]. With the exception of one, all MAH isolates had the same INMV4 pattern. Loci 47, 292, 10, and 7 were monomorphic, whereas locus X3 was the most polymorphic, as previously suggested [Reference Radomski20–22, Reference Inagaki35]. The discriminatory power of the combined MIRU-VNTR was 0·68, lower than those obtained by other researchers 0·80 [Reference Pate22], 0·88 [Reference Radomski20], 0·90 [Reference Inagaki35], and 0·92 [Reference Tirkkonen21]. This finding may be explained by the presence of clones in the concentrated production region of Argentina. Moreover, we identified allelic signatures associated with subspecies of MAC. For instance, MAA and MAS contained allele 1 of locus 25, whereas all MAH and MAP strains contained two or more repeats in this locus [Reference Radomski20, Reference Tirkkonen21, Reference Inagaki35]. Allele 1 was present in all MAA samples, whereas, in MAH strains allele 4 was in locus 25.
As a result of this minimal overlap in the allelic distributions between the different subspecies, MIRU-VNTR-based genotype complexes perfectly correlated with the differentiation of MAH strains from MAA or MAS. Specifically, MAH strains are negative for IS901, whereas MAA or MAS are positive for this sequence. Locus 292 provides information to differentiate between MAP and MAA [Reference Thibault18]. Thus, alleles 3 and 4 of locus 292 were exclusively restricted to, and shared by most of, MAP strains and were absent from the 35 isolates typed in the present study [Reference Thibault18, Reference Radomski20]. This method is technically simple and thus the numerical genotype format may be a very useful tool for molecular characterization of isolates.
The results presented herein demonstrate that the differentiation of MAC strains using MIRU-VNTR is a convenient typing method capable of distinguishing the three main subspecies of the complex and providing new epidemiological knowledge on the strains [Reference Radomski20].
The M. bovis strains identified in co-infection events were spoligotyped and multiple (n = 5) spoligotypes were observed. One of these spoligotypes (SB0140) grouped 80% (16/20) of the isolates and coincided with the most frequent spoligotype identified in cattle from Argentina [Reference Zumárraga36], whereas the remaining spoligotypes (n = 4) only included one isolate each (Table 1).
Co-infection by two different Mycobacteria has been increasingly documented in human TB [Reference Suffys37–Reference de Kantor39], and has incidentally been documented in swine [Reference Agdestein5, Reference de Kantor39, Reference Bartos40]. However, the large proportion (10·2%) of co-infection events detected here suggests that this phenomenon should be further investigated.
This study contributes to the characterization of M. avium strains isolated from pigs in Argentina and, furthermore, is the first report on swine in the country. The relatively low genetic variation and limited spatial extension of MHA isolates suggest that infection is not extensive in the country. Additionally, we reported a proportion of M. avium and M. bovis co-infections larger than those previously reported. These results could contribute to the development of molecular epidemiological tools which may broaden the understanding of disease dynamics in swine. Furthermore, they may help to design and implement effective TB control programmes in Argentina.
ACKNOWLEDGEMENTS
We thank to Dr Julia Sabio y García, Dr Fiorella Kotsias and Dr Maria Mesplet for their critical reading of this manuscript. A.K.G. and A.A.C. are career members of CONICET, Argentina. S.B. holds a fellowship from CONICET, Argentina.
This work was supported by National Institute of Agricultural Technology (AESA 202·831) and UBA (UBACyT 20020100100203) grants.
DECLARATION OF INTEREST
None.





