



Introduction
In 1982, N. Seeman published a landmark theoretical paper that spawned a new field—structural DNA nanotechnology. Reference Seeman1 Motivated by the need to design artificial crystals rationally, Seeman proposed that DNA could be used as a programmable nanoscale building material. Rather than exist simply as isolated double helices, DNA strands could be designed to self-assemble into “branched junctions,” in which several DNA helices came together at a single point. These branched junctions brought DNA into the world of geometric design—they could be combined to fill space with a periodic three-dimensional (3D) latticework, and that latticework could serve as a scaffold to arrange otherwise difficult-to-crystallize proteins so that the protein’s structures could be determined by x-ray crystallography. Seeman’s vision quickly widened to other applications, including a 3D “biochip,” Reference Robinson and Seeman2 which would use metal complexes as the bits of a molecular memory to store an unprecedented four petabytes per cubic centimeter.
As of 2017, the synthesis of crystalline scaffolds for proteins, biochip memories, and other ambitious projects remain incomplete, yet the field of DNA nanotechnology has grown to be practiced by more than 100 labs worldwide and touches fields as different as plasmonics and biophysics. Key current questions include: What advantages does DNA offer over other materials? What challenges limit the widespread use of DNA nanotechnology? And, how might materials scientists accelerate its impact on real-world applications in industry? To address these questions, we begin by comparing DNA to other materials.
The most exciting new materials from the last quarter of a century, from high-temperature superconductors to quantum dots and graphene, have all had signature capabilities—for some physical property these supermaterials exhibit performance that exceeds all others. At first glance, DNA has no such signature capability. Optically, it is a simple UV absorber with technologically unimportant circular dichroism and weak fluorescence—DNA is no competition for tunable and bright quantum dots. Electronically, DNA shows nanometer-scale charge transport via tunneling, Reference Slinker, Muren, Renfrew and Barton3 but this behavior pales in comparison to the micron-scale ballistic transport exhibited by carbon nanotubes and graphene. Catalytically, DNA can be coaxed into cutting other nucleic acids, and its enzymatically gifted cousin RNA likely played a central part in the origin of life, but neither is endowed with the catalytic prowess of protein.
Instead, DNA’s role as stable genetic material, as well as recent interest in its use for mass data storage, Reference Church, Gao, Kosuri and Clelland4 might suggest that DNA’s signature capability is its ability to encode information. However, closer inspection reveals that it is in fact the information processing capabilities of DNA that render it immensely powerful: Solely DNA, RNA, and related unnatural polymers possess the exquisite specificity of Watson–Crick complementarity in which A pairs exclusively with T, and C pairs exclusively with G. This complementarity between strands determines the outcome of information-dependent chemical reactions, whereby two different single-stranded DNA sequences bind strongly and stably if and only if their sequences substantially match. Further, a second, lesser-known information processing operation termed “strand displacement” Reference Zhang and Seelig5 provides a foundation for dynamic DNA-based phenomena: The two DNA strands of one double helix can react with a third DNA single strand to form a new double helix and a released strand, which can then go on to interact with yet another double helix.
Together, these operations enable a myriad of complex static and dynamic molecular systems. For example, via branched junctions and Watson–Crick binding, DNA strands can now be designed to self-assemble into any imaginable shape in both two Reference Rothemund6–Reference Zhang, Jiang, Wu, Li, Mao, Liu and Yan8 and three dimensions. Reference Douglas, Dietz, Liedl, Högberg, Graf and Shih9–Reference Ong, Hanikel, Yaghi, Grun, Strauss, Bron, Lai-Kee-Him, Schueder, Wang, Wang, Kishi, Myhrvold, Zhu, Jungmann, Bellot, Ke and Yin15 DNA strands can also be designed to rearrange themselves via cascades of strand displacement to emulate any temporal dynamic imaginable, Reference Chen, Dalchau, Srinivas, Phillips, Cardelli, Soloveichik and Seelig16 whether that dynamic describes an oscillator, Reference Srinivas, Parkin, Seelig, Winfree and Soloveichik17 an amplifier, Reference Dirks and Pierce18 or a digital circuit. Reference Qian and Winfree19 And the combination of structural and dynamic DNA nanotechnologies offers additional power. When linked to complex DNA geometries, nonequilibrium DNA reactions enable smart containers that use logic to identify cell type Reference Douglas, Bachelet and Church20 to decide when and where to release a drug; bulk hydrogels that melt, solidify, or change shape in response to environmental signals; Reference Previtera, Chippada, Schloss, Yurke and Langrana21–Reference Lu, Guo, Hu, Qi and Willner24 molecular machines that walk bipedally Reference Shin and Pierce25,Reference Omabegho, Sha and Seeman26 like the protein-based molecular motors in our muscles; and the use of these DNA walkers to assemble other nanostructures Reference Gu, Chao, Xiao and Seeman27 or even sort cargo. Reference Thubagere, Li, Johnson, Chen, Doroudi, Lee, Izatt, Wittman, Srinivas, Woods, Winfree and Qian28 Thus, DNA-based materials systems integrate sensing, computation, and actuation of dynamical structure and serve as a universal basis for molecular robotics from the nanometer to the micron scale. Reference Sato, Hiratsuka, Kawamata, Murata and Nomura29
Once developed, complex DNA systems can be used to template other functional materials to take advantage of their signature capabilities. In general, if a material can be chemically conjugated to DNA—and almost any can with sufficient effort—then a DNA structure can both inherit the high-performance properties of that material, as well as offer nanometer-scale organization that enhances performance or even creates entirely new emergent properties. For example, the DNA-directed clustering of plasmonic spheres or rods gives new resonances Reference Fan, He, Bao, Wu, Bao, Schade, Manoharan, Shvets, Nordlander, Liu and Capasso30 or greater fluorescence enhancement Reference Acuna, Möller, Holzmeister, Beater, Lalkens and Tinnefeld31,Reference Puchkova, Vietz, Pibiri, Wünsch, Sanz Paz, Acuna and Tinnefeld32 than observed in isolated, single particles. At a finer scale, DNA can be used to create intricate arrays of dye molecules that exhibit cascades of incoherent energy transfer, Reference Hannestad, Sandin and Albinsson33–Reference Díaz, Buckhout-White, Ancona, Spillmann, Goldman, Melinger and Medintz36 or that coherently couple to produce excitonic circuits that resemble natural light-harvesting systems. Reference Boulais, Sawaya, Veneziano, Andreoni, Banal, Kondo, Mandal, Lin, Schlau-Cohen, Woodbury, Yan, Aspuru-Guzik and Bathe37
The arc of DNA nanotechnology
Technologies and scientific fields have a life history. They are born, undergo growth spurts as intermittent innovations abruptly expand their reach, and eventually mature to a point where they can be used effectively by a wider community as an everyday tool. One example of this, familiar to materials scientists, is semiconductor microfabrication. Its basic techniques are used everywhere from commodity electronics to MEMS accelerometers that trigger airbags and orient cellphones, to microfluidics for creating lab-on-a-chip devices. As a new medium for materials design, DNA nanotechnology appears to be as flexible as microfabrication, and might one day even have as broad a reach.
However, DNA nanotechnology also differs from microfabrication in several important ways. Most prominent among these is that since its inception, microfabrication has been drawn forward by a major commercial need—that of making increasingly complex and powerful electronic circuits. In contrast, DNA nanotechnology has been primarily drawn forward by two forces, neither of which has been a pressing need. The first is a speculative analogy with microfabrication (e.g., Seeman’s biochip). This force is common for “emerging technologies” based on new materials and devices. From carbon-based materials to plasmonics and spintronics, scientists and engineers have appealed to the success and scaling of microelectronics, and held that their new concept could lead analogously to an architecture integrating millions of devices. Each of these emerging technologies is promising and deserves to be explored, and DNA might well play a role in organizing materials for massively integrated device architectures. However, we suggest an alternative, stronger argument for developing DNA nanotechnology.
In particular, we argue that biology provides an “existence proof” for the power of bottom-up self-assembly: It creates molecular machines more intricate than microfabrication has ever achieved; it uses these machines to build everything from bacteria to multicellular organisms; and it drives all of the energetic and material cycles of the biosphere. This argument assumes that research in this direction will necessarily unearth new fundamental principles of self-organization, and it is empowered by the idea that, outside of a potential protein nanotechnology, Reference King, Bale, Sheffler, McNamara, Gonen, Gonen, Yeates and Baker38 there is no existing or imaginable competitor. Within this point of view, it is clear that DNA and RNA have their limits. While special sequences can access Ångstrom-scale structure in the context of aptamers and RNA enzymes, the width of the double helix sets a practical resolution of about 2 nm. And complex 3D structures at the Ångstrom-scale, as well as catalysis, a variety of biological activities, and physical properties such as magnetism or photoluminescence, will likely continue to be the domain of proteins, inorganic nanoparticles, and small molecules. Yet, to take full advantage of their signature capabilities, these materials will continue to be added to DNA, which we suggest will play a central role in programming nanoscale systems at length scales from 2 nm to several microns, and at time scales from minutes to hours. In these spatial and temporal domains, the advantages of engineering with DNA are so great that we predict even advanced protein nanotechnology will not supplant DNA technology. Biology has achieved an advanced nanotechnology using a restricted subset of the periodic table. If we genuinely wish to achieve energy-efficient fabrication of computers, sustainable energy-conversion technologies, and advanced medicine, as we desire, this argument suggests that we must refashion the nanotechnology of biology, potentially using DNA as a starting point.
What is the evidence that we are making progress in this direction? Consider a few results that show how the size and complexity of DNA nanostructures have grown while design and synthesis have become easier. In 1991, Seeman took about one year to construct a 7-nm DNA cube Reference Chen and Seeman39 ( Figure 1a) comprising 10 different short DNA strands and totaling 500 DNA bases in molecular weight. In general, the yield of structures synthesized in this way was highly sensitive to the relative ratios of their component strands, and their design required expert knowledge to create a new shape. By 2006, a different paradigm had emerged, that of “scaffolded DNA origami,” in which a long single “scaffold” strand of DNA was folded and held in place by numerous short DNA “staple” strands. Reference Rothemund6 It enabled the design and synthesis of 2D 100-nm structures (Figure 1b) comprising 200 different strands and 15,000 DNA bases in approximately one week. The self-assembly reaction itself proceeded in a single pot, in a 2 h cooling step with a yield of well-formed structures of more than 95%. Based on a highly regular geometry involving a grid of branched junctions that arrayed helix axes in simple parallel arrays, DNA origami was highly insensitive to the relative ratio of components, easy to generalize, and in a single paper, one-half dozen unique shapes and another one-half dozen distinct patterns were reported.
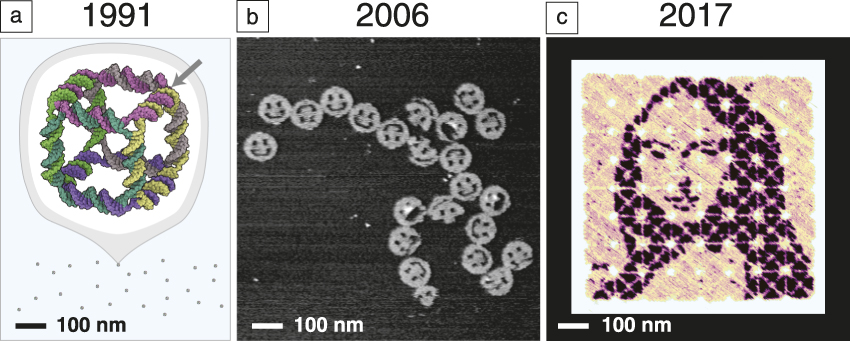
Figure 1. Snapshots from the evolution of DNA nanotechnology. (a) Seeman’s original DNA cube, constructed from 10 strands using hybridization and enzymatic ligation over five steps to give a 170 kilodalton molecular complex in 1% yield by gel. The arrow indicates one of 12 three-arm branched junctions at the cube’s vertices. Reprinted with permission from Reference Reference Chen and Seeman39. © 1991 Macmillan Publishers Ltd. (b) An early DNA origami design, constructed from 244 strands using only a single step of DNA hybridization to give a 4.7 megadalton complex in 72% yield by atomic force microscopy. Reference Rothemund6 (c) State-of-the-art hierarchical assembly of 64 DNA origami squares from 394 strands over four steps (hybridization and mixing) to give a ∼400 megadalton complex in 3% yield. Adapted with permission from Reference Reference Tikhomirov, Petersen and Qian43. © 2017 Macmillan Publishers Ltd. This structure bears a pattern encompassing 8704 independent pixels with 6-nm resolution. A major challenge, for which the field has several potential solutions, is how to transfer such 2-nm-thick patterns into a high-performance functional material.
Extensions to 3D architectures quickly followed. Reference Douglas, Dietz, Liedl, Högberg, Graf and Shih9 The size of an individual origami is fundamentally limited by the length of the long single-stranded “scaffold” from which it is folded. Various approaches to creating longer scaffolds, Reference Marchi, Saaem, Vogen, Brown and LaBean40 combining origami, Reference Rajendran, Endo, Katsuda, Hidaka and Sugiyama41,Reference Woo and Rothemund42 or returning to scaffold-less “single-stranded tile” paradigms Reference Wei, Dai and Yin7,Reference Ke, Ong, Shih and Yin12,Reference Ong, Hanikel, Yaghi, Grun, Strauss, Bron, Lai-Kee-Him, Schueder, Wang, Wang, Kishi, Myhrvold, Zhu, Jungmann, Bellot, Ke and Yin15 (SST, using only short strands) have all been successful in increasing the size of DNA structures. One approach Reference Tikhomirov, Petersen and Qian43 combines 64 square origami tiles into a 700 nm × 700 nm array (Figure 1c). Importantly, every single 6-nm “pixel” in such a structure can be toggled on and off, and an arbitrary pattern can be self-assembled. The hierarchical nature of the approach also makes the most efficient use of DNA synthesis—the same 394 strands are used over and over to make each of the 64 tiles, and thus the synthetic cost is little more than that required to create a single-origami pattern. The resulting pattern features 8704 pixels encompassing almost one million DNA bases and, in just 30 min, an automatic pipetting robot can mix up a new pattern for any desired image. This advance, and others in computer-aided design, Reference Douglas, Marblestone, Teerapittayanon, Vazquez, Church and Shih44 hands-free shape-to-sequence automated design, Reference Benson, Mohammed, Gardell, Masich, Czeizler, Orponen and Högberg13,Reference Veneziano, Ratanalert, Zhang, Zhang, Yan, Chiu and Bathe14 structure prediction and mechanical analysis via coarse-grained simulation Reference Kim, Kilchherr, Dietz and Bathe45–Reference Pan, Bricker, Ratanalert and Bathe47 or molecular dynamics, Reference Pan, Bricker, Ratanalert and Bathe47,Reference Yoo and Aksimentiev48 experimental characterization or control over folding rate Reference Sobczak, Martin, Gerling and Dietz49 and elucidation of the fundamental mechanisms Reference Dunn, Dannenberg, Ouldridge, Kwiatkowska, Turberfield and Bath50–Reference Lee Tin Wah, David, Rudiuk, Baigl and Estevez-Torres53 of DNA nanostructure formation, along with practical and cost-effective techniques for their milligram- Reference Kick, Praetorius, Dietz and Weuster-Botz54 to gram- Reference Praetorius, Kick, Behler, Honemann, Weuster-Botz and Dietz55 scale synthesis are making structural DNA nanotechnology increasingly accessible. If a researcher does not wish to design a structure themselves, one company 56 even offers design and synthesis services for custom DNA origami.
A similar amount of progress has been made in dynamic DNA technology triggered by the demonstration of a DNA tweezer, Reference Yurke, Turberfield, Mills, Simmel and Neumann57 which could be cyclically opened and closed by the manual addition of DNA fuel strands. Such systems have transitioned from manual to autonomous actuation, as the field has mastered the synthesis of metastable fuels. The state of the art is best captured by a three-phase autonomous oscillator, Reference Srinivas, Parkin, Seelig, Winfree and Soloveichik17 and the result that any behavior that can be expressed as a set of chemical reactions can be realized with DNA dynamics. Reference Chen, Dalchau, Srinivas, Phillips, Cardelli, Soloveichik and Seelig16 On the practical side, dynamic DNA systems enable new types of super-resolution microscopy Reference Jungmann, Steinhauer, Scheible, Kuzyk, Tinnefeld and Simmel58,Reference Jungmann, Avendaño, Woehrstein, Dai, Shih and Yin59 and imaging of biological tissue Reference Choi, Chang, Trinh, Padilla, Fraser and Pierce60 with a high level of multiplexing. Kits and reagents enabling these application are also available from both nonprofit academic organizations 61 and recent startup companies. 62
Despite its compelling prototypes and promise, widespread adoption of DNA as a programmable medium has been slow. Of the many factors that control its diffusion as a technology, we describe two that we believe are most important. First, structural and dynamic DNA nanotechnology are neither conceptually complete nor technologically “plug-and-play.” At a fundamental level, questions such as how to predict the melting temperature of DNA origami or SST structures remain unanswered. Most practitioners of DNA origami use suboptimal natural sequences as scaffolds and most SST designs use random sequences. Consequently, another major theoretical question is whether artificial sequences that are optimal relative to yield or defect density can be designed.
At a practical level, even if ideal artificial sequences can be designed, there are still challenges. Efforts to scale up gene synthesis continue to make the synthesis of larger structures more cost effective, but most techniques produce double-stranded DNA, and hence the problem of creating single strands is a highly active area of research. Reference Veneziano, Ratanalert, Zhang, Zhang, Yan, Chiu and Bathe14,Reference Zhang, Chao, Pan, Liu, Huang and Fan63,Reference Erkelenz, Bauer, Meyer, Gatsogiannis, Raunser, Saccà and Niemeyer64 Cost remains an issue where large amounts of nanostructures are required (e.g., for gels, Reference Bertrand, Fygenson and Saleh23 liquid crystals, Reference Douglas, Chou and Shih65,Reference Siavashpouri, Wachauf, Zakhary, Praetorius, Dietz and Dogic66 or potential therapeutics Reference Douglas, Bachelet and Church20 ); at the scale achievable in most labs, DNA origami costs ∼USD$20,000 per gram. Currently, DNA staple strands and SST strands are typically produced via costly solid-state chemical synthesis. For DNA structures to serve as a basis for bulk materials, liquid-state chemical synthesis or biological Reference Praetorius and Dietz67 production in bacteria (∼USD$200/g of DNA origami at large scale) will need to be further developed. For applications where chemical modifications are used to provide function, more cost-effective means of solid-state or liquid-state chemical and enzymatic synthesis Reference Veneziano, Ratanalert, Zhang, Zhang, Yan, Chiu and Bathe14,Reference Ducani, Kaul, Moche, Shih and Högberg68 are needed.
Sequence design and synthesis are only half of the story. The field’s eventual goal is to completely automate the process of design, so that a high-level concept for a device can be specified abstractly and then geometric and sequence design can proceed without further human intervention. Just as a solid object can now be scanned with a cell phone, converted to a model, and 3D printed, the same workflow should eventually be possible for molecular assembly. For mesh-based designs that are suitable for rendering polyhedra and any structure that can be approximated by polygons, this goal has been realized, Reference Benson, Mohammed, Gardell, Masich, Czeizler, Orponen and Högberg13,Reference Veneziano, Ratanalert, Zhang, Zhang, Yan, Chiu and Bathe14 but for structures constructed from arrays of parallel helices, Reference Rothemund6,Reference Douglas, Dietz, Liedl, Högberg, Graf and Shih9 considerable computer-aided handicraft is still required. Pixel-based architectures, such as the hierarchical origami approach Reference Tikhomirov, Petersen and Qian43 previously described and SST, Reference Wei, Dai and Yin7,Reference Ke, Ong, Shih and Yin12,Reference Ong, Hanikel, Yaghi, Grun, Strauss, Bron, Lai-Kee-Him, Schueder, Wang, Wang, Kishi, Myhrvold, Zhu, Jungmann, Bellot, Ke and Yin15 provide the ability to rapidly print approximations of arbitrary 2D patterns or 3D shapes, but such approximations necessarily conform to a grid, which limits the design of mechanical devices. A single unified design environment for completely general DNA geometries has yet to be developed. Further, combining low-cost, large-scale synthesis, as envisioned, together with fully automated in silico design of custom functional materials Reference Sun, Boulais, Hakobyan, Wang, Guan, Bathe and Yin69 such as plasmonic nanoparticles, is the holy grail of DNA-based materials production. This vision is in line with the Materials Genome Initiative, which seeks to enable the in silico design and analysis of functional materials for their high-throughput, rapid discovery, and development. Reference Jain, Ong, Hautier, Chen, Richards, Dacek, Cholia, Gunter, Skinner, Ceder and Persson88
With respect to geometry, there is significant research into basic architectures. A long-standing dream has been to create wholly single-stranded designs that fold similarly to the single amino acid chain of a protein: entirely intramolecularly, during polymerization, at a single physiological temperature. Such an architecture could be genetically encoded and cloned, simultaneously enabling (1) large-scale biological production, (2) mutation and in vitro selection of structures to harness the power of evolution to add function, and (3) in situ use of the nanostructures within cells to scaffold enzymes or perhaps even create new classes of artificial organelles. In this direction, a major milestone was reached in 2004 with the publication of a 1700-base single-stranded DNA octahedron Reference Shih, Quispe and Joyce70 that used just a handful of shorter strands to rigidify the structure. Entirely single-stranded RNA architectures have been synthesized isothermally, Reference Geary, Rothemund and Andersen71 and larger single-stranded paranemic-crossover-based DNA and RNA structures have been synthesized via thermal annealing. Reference Han, Qi, Myhrvold, Wang, Dai, Jiang, Bates, Liu, An, Zhang, Yan and Yin72 Yet, no single-stranded architecture has simultaneously achieved large size, high complexity, isothermal folding, and expression in living cells.
A second major barrier to the diffusion of DNA nanotechnology is the challenge of building good interfaces and achieving integration with respect to different materials systems, classical microfabrication, and biology. Simply coupling DNA to a new type of nanoparticle might be chemically straightforward, but rigidly attaching exactly one functional component to one DNA structure in a desired orientation may be challenging. Assuming that a functional material is perfectly organized by DNA to create a device, delivering that device to its point of use is an equal and often underappreciated challenge. Solution-synthesized DNA structures deposit randomly on chips, and creating connections to individual devices with standard techniques like e-beam lithography is unscalable. Analogously, solution-synthesized DNA devices that perform well in vitro often cannot be efficiently delivered to the right compartment within cells, or may degrade rapidly within an organism. Potential solutions to solid-state integration Reference Gopinath and Rothemund73 and biological delivery Reference Perrault and Shih74,Reference Kiviaho, Linko, Ora, Tiainen, Järvihaavisto, Mikkilä, Tenhu, Nonappa and Kostiainen75 are being explored, but there is no shortage of such challenges, and general solutions will require major conceptual advances. Materials scientists have long specialized in building artful interfaces Reference Dunlop, Weinkamer and Fratzl76 between seemingly incompatible materials, so they are perfectly positioned to bridge the DNA world with the greater materials universe.
In this issue
DNA nanotechnology already has deep roots in materials science ( Figure 2 ). Its branches encompass many areas of interest to the materials science community (e.g., nanophotonics, lithography, and metrology) as well as a few that might be unfamiliar (e.g., enzyme cascades and RNA scaffolds). The 10 articles in this issue of MRS Bulletin aim to introduce materials scientists to the major branches of DNA nanotechnology.
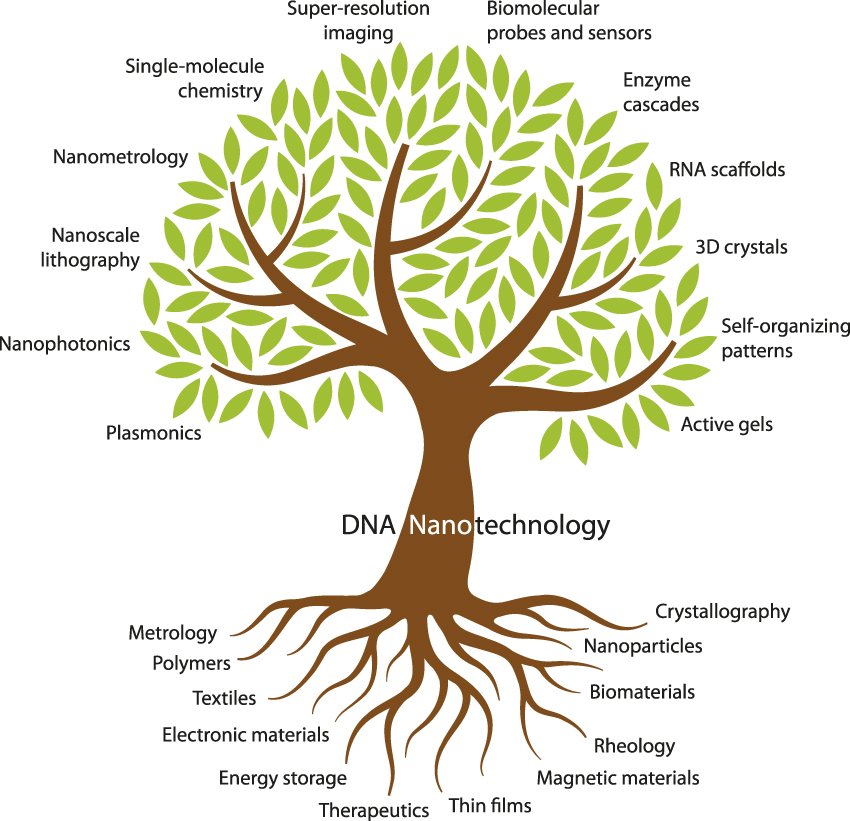
Figure 2. Fundamental topics and tools of materials science (bottom) provide support for the various subdisciplines of DNA nanotechnology (top) covered in this issue.
The issue leads off with an article by Wang et al., Reference Wang, Chatterjee, Yan, LaBean, Turberfield, Castro, Seelig and Ke77 which presents an introduction to programming structural and dynamic nucleic acid systems for anyone new to the field. An article by Gothelf Reference Gothelf78 follows with a review on the intersection of chemistry and DNA nanotechnology. The article covers both the basic chemical modification of DNA, which underlies the interface problem of coupling diverse materials to DNA, as well as advanced applications, such as the photoswitching of azobenzenes to add motion to DNA machines, or DNA templates programmed to control the outcome of chemical reactions.
In their article, Seeman and Gang Reference Seeman and Gang79 collaborate to take us into the third dimension. They provide both an update on Seeman’s original research program to synthesize high-quality 3D crystals from DNA-only building blocks, to use as hosts for proteins or potential memory molecules, as well as a review of how the principles of 3D self-assembly fundamentally change when nanoparticle-DNA hybrids are used as building blocks. Simmel and Schulman Reference Simmel and Schulman80 lead a tour of the many ways that DNA systems can be used to self-organize patterns, from discrete complexes that assemble into nanometer-scale fractal tapestries, to bulk reactions whose continuum dynamics drive millimeter-scale waves. They highlight connections to soft matter, including the compartmentalization of reactions within vesicles, and the programmability that DNA brings to hydrogels. This relatively unexplored area, where nanoscale DNA structure affects bulk materials properties, is ripe for materials scientists wishing to explore the fundamental relationship between geometry and gel properties, as well as the medical application of biocompatible gels whose stiffness and density can be switched at will.
Turning away from general explorations of DNA’s organizational power to applications involving specific functional materials, an article by Grossi et al. Reference Grossi, Jaekel, Andersen and Saccà81 illustrates how DNA can achieve exquisite control over protein enzymes. This includes the organization of multiple enzymes into cascades that act as nanoscale factories, where the product of one reaction is handed off from one enzyme to another in an assembly line, as well as clever artificial devices, such as a box encapsulating an enzyme with a lid that controls the enzyme’s access to substrate molecules and thus controls reaction rate. A major open problem for DNA-based enzymatic control is to reach the performance, in terms of rate enhancement or specificity, of natural protein-scaffolded multi-enzyme systems.
In their article, Castro et al. Reference Castro, Dietz and Högberg82 expand on the theme of artificial DNA devices, with an emphasis on using mechanical devices to study proteins or other molecules, measuring their interaction energies, intermolecular distances, or the forces between them. For the practicing biologist or biophysicist, this amounts to a new paradigm for answering scientific questions. Instead of figuring out which existing tools can be used to indirectly deduce a property of molecules, the experimenter designs a “custom DNA origami instrument” whose geometry and mechanics allow the direct measurement of the property of interest.
DNA-based devices form and perform beautifully in vitro, but there are obstacles to their use within living cells. DNA nanostructures are typically synthesized via a thermal ramp that starts at a temperature too high for most cells to survive, reliably targeting them to a desired cellular compartment is difficult, and the usual chemical methods for integrating proteins with DNA may fail inside cells. To provide a route to intracellular devices ranging from enzyme scaffolds to custom instruments, new isothermally folded, genetically encodable RNA architectures are being explored. Created by cells themselves, these structures will more easily interface with proteins via a wealth of natural RNA sequences that bind to protein. RNA nanostructures have the further advantage that they can be enzymatically synthesized at milligram scale in vitro, and perhaps grown at even greater scales in cells. In their article, Weizmann and Andersen Reference Weizmann and Andersen83 discuss such structures, which present new challenges. For example, RNA structures can easily become stuck in topologically frustrated kinetic traps during folding, and thus new methods are needed to understand knotted RNAs.
More classical applications of materials science are emphasized in the final trio of articles, which reflect one of the most interesting recent trends—an increasing focus on optical devices, measurements, and integration. Since Seeman’s biochip proposal, DNA nanotechnology has been suggested as a route to fabricating nanoelectronics, but the creation of reproducible electrical connections to self-assembled nanodevices continues to be an open problem. Optical applications sidestep this challenge, as most do not depend on nanometer-scale connections. Spotlighting this theme, Pilo-Pais et al. Reference Pilo-Pais, Acuna, Tinnefeld and Liedl84 discuss the wide range of effects that can be achieved by using DNA nanostructures to organize plasmonic nanoparticles and light emitters such as dyes and quantum dots. For sensing applications, DNA can precisely position single analyte molecules between nanoparticle dimers to achieve extraordinary enhancement of fluorescence signals; for metamaterials, DNA offers large-scale synthesis of metal nanoparticle crystals, and can achieve large chiral optical responses, in which the handedness of light is switched by the handedness of the DNA template.
In their article, Xu et al. Reference Xu, Harb, Kostiainen, Hughes, Woolley, Liu and Gopinath85 focus on DNA nanofabrication. The authors envision DNA origami as breadboards upon which diverse components can be assembled, and address the often-neglected micron-scale integration of the breadboards into conventionally microfabricated devices. More conventional fabrication paradigms are also explored, including the use of DNA structures as etch masks for semiconductors, as growth sites for oxides or metals, and as masters for nanoimprint. This highlights another important challenge for materials scientists—the problem of faithfully transferring fine details of DNA structures into high-performance materials needs more and better solutions. The authors end with two proposals emphasizing DNA nanofabrication’s unique benefits—the self-assembly of 3D circuits and 2D patterning for optical metasurface construction over large areas at low cost.
Concluding the issue, Graugnard et al. Reference Graugnard, Hughes, Jungmann, Kostiainen and Linko86 explore how dynamic and structural DNA nanotechnology can be used cooperatively to enable both new types of super-resolution imaging (DNA-PAINT), and calibration of super-resolution microscopes. Notably, the use of DNA origami rulers as calibration standards is the first commercial application of DNA nanostructures. 87 The authors end with a creative proposal for how DNA nanostructures might provide the foundation for a sub-10-nm lithography in which defects are optically self-documenting.
We have seen that DNA’s signature capability is information processing, which enables both complex spatial organization and dynamical reconfiguration of high-performance functional materials. On its own, the field of DNA nanotechnology will continue to push the limits of DNA’s signature capability and will seek to translate its prototypes and demonstrations into real-world applications. But with more interest from mainstream materials scientists, progress will be greatly accelerated. Personally, we are fascinated by the versatility of DNA as an information processing polymer to autonomously shape itself into complex molecular machines, and to take on and improve the functionality of the other materials that it organizes. We hope that this issue of the MRS Bulletin will instill the same fascination in its readers, whom we invite to engage in the pursuit of using DNA as a programmable engineering material.
Acknowledgments
M.B. acknowledges the Office of Naval Research (N00014-16-1-2181, N00014-13-1-0664, N00014-15-1-2830, N00014-16-1-2506, N00014-16-1-2953, N00014-17-1-2609); the National Science Foundation (CMMI-1334109, CCF-1547999, CCF-1564025); the Army Research Office (W911NF-12-1-0420); the US Department of Energy (DE-SC0001088; DE-SC0016353); the National Institutes of Health (U01-MH106011; R01-MH112694); and the Human Frontier Science Program (RGP0029/2014) for funding to support DNA-related research in his group. P.W.K.R acknowledges the Office of Naval Research (N00014-14-1-0702, N00014-16-1-2159); National Science Foundation (CCF-1317694, CCF-0832824, CMMI-1636364); Army Research Office (W911NF-11-1-0117); Microsoft Research; the Semiconductor Research Corporation (FENA); the Orr Family Foundation; and Abedin Institute for funding his group’s DNA nanotechnology research.

Mark Bathe is an associate professor in the Department of Biological Engineering at the Massachusetts Institute of Technology (MIT); an associate member of the Broad Institute of MIT and Harvard University; and a member of the MIT Center for Excitonics, the MIT Center for Neurobiological Engineering, and the MIT Center for Environmental Health Sciences. He obtained his bachelor’s, master’s, and doctoral degrees at MIT working in the Departments of Mechanical Engineering, Chemical Engineering, Chemistry, and Biological Engineering. He carried out his postdoctoral research in biological physics at the University of Munich, Germany, returning to MIT in 2009 to join the faculty in the Department of Biological Engineering. His current research focuses on developing integrated computational-experimental approaches for engineering biology. Bathe can be reached by email at [email protected].

Paul W.K. Rothemund is a research professor at the California Institute of Technology (Caltech) in the Departments of Bioengineering, Computing + Mathematical Sciences, and Computation & Neural Systems. He is also a graduate of Caltech, where he dually majored in biology and engineering and was awarded the first patent for a DNA computer. He received his PhD degree in theoretical computer science from the University of Southern California. He is the inventor of DNA origami, and his current research includes the integration of DNA origami with microfabrication and the design of isothermally folded RNA origami, which may be genetically encoded to create scaffolds for ribonucleoprotein machines. He is the recipient of a Feynman Prize, World Technology Award, and MacArthur Fellowship. Rothemund can be reached by phone at 626-390-0438 or by email at [email protected].


