


INTRODUCTION
Tuberculosis (TB) is the most prevalent, but preventable infectious disease worldwide, accounting for 9·2 million new cases and nearly 1·7 million deaths (about 25% of all preventable deaths) annually [1]. Developing countries bear over 90% of all global cases, and China has the second highest TB burden in the world. TB is also the most common and fatal infectious disease in China, accounting for 43% of deaths from infectious diseases [Reference Wang, Liu and Chin2]. Over the last decade, drug-resistant TB has been emerging as a major concern in China [Reference Wang, Zhang and Duanmu3, 4]. A national survey of TB in 2000 reported that 27·8% of Chinese TB patients were resistant to at least one of the first line anti-TB drugs, with 10·7% resistant to more than one anti-TB drugs [5]. China has the highest burden of drug-resistant TB in the world and accounts for a quarter of the global burden [4, Reference Liu6, Reference Zignol7]. The pandemic of drug-resistant TB in China was proven to be related to incomplete anti-TB chemotherapy due to unsatisfactory patient compliance and the poor implementation quality of directly observed treatment, short course (DOTS) [Reference Liu8].
In addition to incomplete anti-TB chemotherapy, molecular epidemiological studies have suggested that associations between certain genotypes (strains) of M. tuberculosis and the drug-resistant phenotypes may exist. Of these, the Beijing family, a widespread and dominant M. tuberculosis genotype in China and throughout Asia, has been extensively investigated in a variety of geographic regions and populations [Reference van Soolingen9–Reference Hu11]. In some studies, the Beijing family genotype exhibited important pathogenic features associated with high virulence and multidrug resistance (MDR) rate [Reference Park12, Reference Toungoussova13]. But these findings were not confirmed by other studies [Reference Glynn14]. Additional genotyping strategies are required to refine the Beijing family and identify possible subgroups, which may explain the drug resistance scenario. A recent study conducted in the Shandong Province of China, has shown that 81% of M. tuberculosis strains in this province were members of the Beijing family [Reference Ma15]. Beijing family isolates were found more often among rifampicin (RIF)-resistant isolates (90·0%) than among RIF-susceptible isolates (82·7%), but the difference was not statistically significant [Reference Ma15]. Meanwhile, a major subgroup of Beijing family has been identified with 12-loci mycobacterial interspersed repetitive units (MIRU) genotype 223325173533, which accounts for 30% of Beijing family isolates in Shandong Province [Reference Ma15]. Importantly, this genotype was found significantly more often among RIF-resistant isolates than among RIF-susceptible isolates (41·7% vs. 25·3%, P=0·04) [Reference Ma15]. Subsequently, the US Centers for Disease Control and Prevention (CDC) has recognized the importance of this genotype and designated it as PCR00002 based on the two PCR genotyping methods (spoligotyping and MIRU) in the TB Genotyping Information Management System (TB GIMS). CDC strain 210, a widely distributed Beijing family strain in the USA, is also a member of this major subgroup. These data suggest the importance and necessity of further investigating this genotype and its association with drug resistance in different populations and geographic regions. In the current study, we analysed the molecular characteristics of drug-resistant M. tuberculosis through a population-based molecular epidemiological study of TB conducted in two rural counties from two provinces (Zhejiang Province and Jiangsu Province) of eastern China. Our data provide additional evidence for the association between this major subgroup of Beijing family and MDR phenotypes.
METHODS
Study population
A population-based epidemiological study of active TB was conducted in two rural counties of eastern China, Guanyun County in Jiangsu Province (between 1 April 2004 and 31 March 2005) and Deqing County in Zhejiang Province (between 1 June 2004 and 31 May 2005). All pulmonary TB cases identified in these two counties within the study period were enrolled in this study. Sputum specimens from these patients were submitted to the microbiology laboratory at the School of Public Health, Fudan University for specimen processing, M. tuberculosis culture, identification, drug susceptibility testing, DNA sequencing and genotyping. A total of 351 M. tuberculosis isolates (187 from Guanyun County and 164 from Deqing County) were successfully collected from the enrolled patients, including 258 (91·2%) from new TB cases and 93 (80·2%) from re-treated TB cases.
Drug susceptibility testing
Drug susceptibility testing for the four first line anti-TB drugs [isoniazid (INH), rifampicin (RIF), ethambutol (EMB), and streptomycin (SM)] was performed by the proportion method. The final concentrations of these four drugs in Löwenstein–Jensen culture were 0·2, 40, 2 and 10 mg/l, respectively. A positive result was defined by the growth of >1% of colonies compared to the drug-free control.
Molecular characterization
As the study objective was to further investigate the association between subgroups of Beijing family and drug resistance phenotype, three genotyping methods were applied to molecularly characterize these M. tuberculosis isolates: spoligotyping, MIRU genotyping, and IS6110 restriction fragment length polymorphism (RFLP) genotyping. Extraction of M. tuberculosis genomic DNA was performed by standard methods [Reference van Embden16]. Spoligotyping was performed by using a commercial kit from Isogen Bioscience BV (Maarssen, The Netherlands) [Reference van Embden16]. Strain family and clade similarity were determined by comparison of their spoligotyping pattern with the SpoligDB4 database using the SPOTCLUST program available online (http://cgi2.cs.rpi~bennek/SPOTCLUST.html). Beijing family M. tuberculosis was defined as a strain that hybridized to only the last nine spacer oligonucleotides (spacers 35–43) of the spoligotyping pattern. MIRU (12 loci) genotyping was applied to identify the subgroup of the Beijing family, and was performed by PCR amplification of the 12 MIRU loci (MIRU2, 4a, 10, 16, 20, 23, 24, 26, 27, 31b, 39, 40) following the protocol described previously [Reference Cowan17]. Each strain was given a 12-digit MIRU identification number. The allelic diversity and discriminatory power of MIRU loci were calculated using the h value and Hunter–Gaston index (HGI), respectively, as previously reported [Reference Selander18, Reference Hunter and Gaston19]. IS6110 RFLP was conducted by following the standard protocol of van Embden et al. [Reference van Embden16], with data analysed by Gelcompar II (Applied Maths, Belgium). A molecular ‘cluster’ was defined as M. tuberculosis isolates from two or more patients having an identical IS6110 RFLP pattern. Typically, ‘clustered’ strains indicate recent transmission while ‘unique’ strains indicate reactive disease from a remote infection.
Hotspot mutations within the katG and rpoB genes of drug-resistant isolates were analysed by PCR direct sequencing. Primers pairs were CCCATGGCCGCGGCGGTCGACATT and CGCCGTCCTTGGCGGTGTATTGCC for katG gene (Accession no. X68081), and GGGAGCGGATGACCACCCA and GCGGTACGGCGTTTCGATGAAC for rpoB gene (Accession no. L27989). Mutations in these genes were determined by amplification of the corresponding hotspot mutation region by PCR using GeneAmpPCR System 9700 (Applied Biosystems Inc., USA). The PCR reactions followed the manufacturer's protocol of HotStar Taq DNA polymerase (Qiagen Inc., Germany). The full-length sequencing of targeted PCR products were performed by an ABI 3770 DNA sequencer (Applied Biosystems Inc.). Raw sequencing data was carefully analysed by Sequencher 4.7 software (Gene Codes Corporation, USA) and independently confirmed by two molecular biologists.
Statistical analysis
The χ2 test was used to assess variations in clustering proportion of M. tuberculosis isolates with different genotypes and phenotypes of drug resistance. Fisher's exact two-tailed test was used when the expected value was <5. In the binary logistic model, the odds ratio for clustering proportion was adjusted for age, sex, and county of the subjects. Statistical analysis was by SPSS software 11.0 (SPSS Inc., USA).
RESULTS
Of the 351 M. tuberculosis isolates collected from the two counties, 223 (63·5%) were resistant to at least one anti-TB drug, including 147 (57·0%) isolates from 258 new TB cases and 76 (81·7%) from 93 re-treatment TB cases. Compared with the new TB cases, the re-treated TB cases were more likely to develop any drug resistance (P<0·01), mono-INH resistance (P=0·01), MDR (resistant to at least INH and RIF, P=0·04), and resistance to RIF and SM (P=0·04) (Table 1).
Table 1. Characteristics of M. tuberculosis isolates from Deqing and Guanyun counties
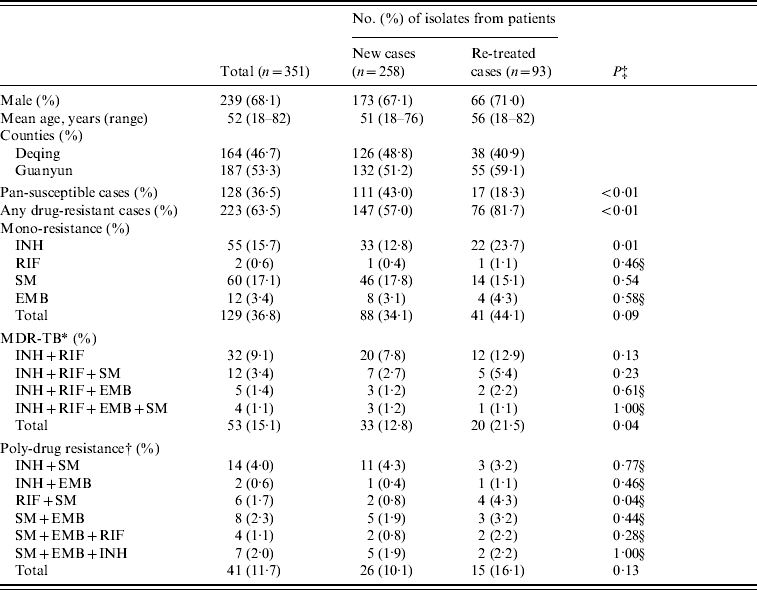
INH, Isoniazid; RIF, rifampicin; EMB, ethambutol; SM, streptomycin.
* Defined as resistant to at least INH and RIF.
† Defined as resistant to at least two drugs except MDR.
‡ P value by the χ2 test.
§ P value by Fisher's exact test.
Spoligotyping identified 243 (69·2%) isolates with the Beijing family genotype, 35 (10·0%) with the family 33 genotype, 21 (6·0%) with the T lineage genotype, seven (2·0%) with the Haarlem genotype, seven (2·0%) with the EAI genotype, and five (1·4%) with the LAM genotype. Thirty-three (9·4%) isolates had unique/novel spoligotypes, of which similarity analysis found that 19 isolates shared >50% similarity with family 33, 12 similar to the T lineage, and two similar to the Haarlem genotype.
Twelve MIRU loci showed varied heterogeneity from MIRU26 (h=0·76) to MIRU2 (h=0·003). MIRU genotyping alone identified 235 genotypes (HGI=0·9317), including 189 ‘unique’ patterns and 46 ‘cluster’ patterns containing 162 isolates. MIRU genotyping refined the Beijing family into 139 different MIRU genotypes (HGI=0·9232) and identified 96 MIRU genotypes among non-Beijing family isolates (HGI=0·9874). No MIRU genotype was observed in both Beijing family and non-Beijing family isolates. The largest MIRU group contained 38 isolates with identical MIRU genotype 223325173533, all of which belonged to the Beijing family and accounted for 15·6% of Beijing family isolates in this study. This major subgroup of the Beijing family shared the same definition with US CDC PCR00002 genotype (Beijing family plus MIRU 223325173533).
Each MIRU-defined subgroup was further analysed by IS6110 RFLP. The isolates in the major subgroup (PCR00002) presented more IS6110 copy numbers (mean 18, range 13–23) on average than other isolates of the Beijing family (mean 16, range 8–23) and non-Beijing family isolates (mean 10, range 2–17). IS6110 RFLP identified 22 clusters (66 isolates) in the Beijing family and six clusters (14 isolates) in the non-Beijing family; no IS6110 RFLP pattern was shared between Beijing and non-Beijing families. The size of clusters ranged from 2 to 6 isolates in the Beijing family and from 2 to 4 isolates in the non-Beijing family, respectively. Twenty-three (60·5%) of 38 isolates in the major subgroup (PCR00002) were identified within ten IS6110 RFLP clusters (Fig. 1).

Fig. 1. Cluster analysis of the major subgroup (MIRU genotype, 223325173533/PCR00002) of Beijing family. * Drug resistance pattern sequence: isoniazid, rifampicin, ethambutol, streptomycin, respectively. R, Resistant; S, susceptible.
Hotspot mutations in the katG and rpoB genes associated with INH and RIF resistance, respectively, were identified by re-sequencing of the genes in all 351 M. tuberculosis isolates (Table 2). Beijing family isolates contained more mutations in the katG gene (29·2% vs. 9·3%, P<0·01) and rpoB gene (21·0% vs. 8·3%, P<0·01), respectively, compared to non-Beijing family isolates. Within Beijing family, the isolates in the major subgroup (PCR00002) carried significantly more mutations in the katG gene (57·9% vs. 23·9%, P<0·01) and rpoB gene (44·7% vs. 16·6%, P<0·01) compared to other Beijing family isolates. In the major subgroup (PCR00002), 19/23 clustered isolates contained the mutation in codon 315 of the katG gene, with most of mutation being katG315Leu and significantly more observed than the unique isolates (82·6% vs. 13·3%, P=0·0176).
Table 2. The position and frequency of genetic mutation associated with drug-resistant TB
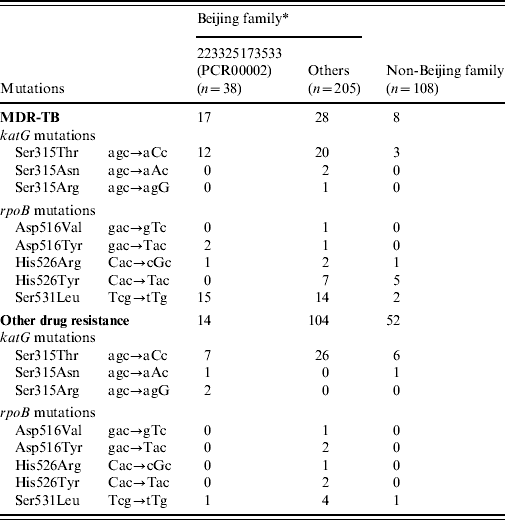
* Within the Beijing family, One MDR isolate and one other drug-resistant isolate presented multiple mutations of rpoB516GTC(Val) and rpoB531TTG(Leu); One MDR-TB isolate presented rpoB516TAC(Tyr) and rpoB531TTG(Leu) simultaneously.
A binary logistic regression model adjusted for the age, sex, and county of subjects was applied to analyse the associations in genotypes (i.e. Beijing family and its major subgroup), drug-resistance phenotypes, gene mutations and IS6110 RFLP-defined clustering (Table 3). The Beijing family isolates presented increased risks for being MDR [18·5% vs. 7·4%, odds ratio (OR) 3·02, 95% confidence interval (CI) 1·35–6·73] and having katG and rpoB mutations (12·8% vs. 3·7%, OR 3·81, 95% CI 1·30–11·09) compared to non-Beijing family isolates. Cluster analysis by IS6110 RFLP revealed that Beijing family isolates had an increased risk for being clustered (27·2% vs. 13·0%, OR 2·65, 95% CI 1·39–5·04) compared to non-Beijing family isolates. Within the Beijing family, the isolates in the major subgroup (PCR00002) showed significantly higher risks for MDR (44·7% vs. 13·7%, OR 6·18, 95% CI 2·68–14·23), having katG and rpoB mutations (31·6% vs. 9·3%, OR 4·27, 95% CI 1·86–9·80), and being clustered (60·5% vs. 21·0%, OR 6·14, 95% CI 2·82–13·37) compared to other isolates in the Beijing family. In addition, the isolates in the major subgroup (PCR00002) were found more often in re-treated cases than other isolates of the Beijing family (39·5% vs. 25·0%, P=0·04).
Table 3. Association between M. tuberculosis genotypes and drug-resistance phenotypes/gene mutations/clustering
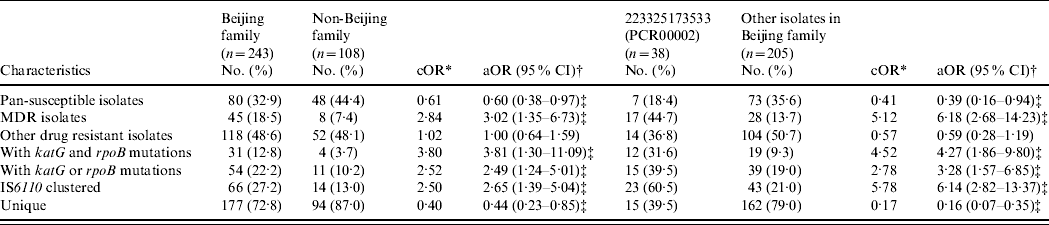
* cOR, crude odds ratio was calculated by comparing the variables between Beijing genotype and non-Beijing genotype, or between PCR00002 and other isolates in Beijing family from χ2 test.
† aOR, adjusted odds ratio was calculated by comparing variables between Beijing genotype and non-Beijing genotype, or PCR00002 and other isolates in Beijing family, adjusted by age, sex, and counties of the subjects in a binary logistic regression model.
‡ P<0·05.
Contact investigation using the concentric circle method (household, working, social contacts) was conducted to investigate the epidemiological links in patients infected with the MIRU/IS6110 RFLP clustered isolates. The confirmed epidemiological links were identified from 26 (32·5%) of 80 patients infected with clustered isolates. Furthermore, five (21·7%) of 23 patients infected with the clustered major genotype (PCR00002) isolates were found to have epidemiological links.
DISCUSSION
MDR-TB has become a major public health concern in China, predominantly occurring in resource-limited rural areas. To date, there has been no large-scale population-based epidemiological study investigating molecular characteristics of MDR-TB strains in rural China. Our study was based on a population-based epidemiological study of TB in the Chinese populations living in two rural counties of eastern China, which had an overwhelmingly high prevalence of MDR-TB of 18·7% and 11·0%, respectively [Reference Hu20]. In addition to socio-economic factors causing inefficient implementation of DOTS strategy [Reference Xu21, Reference Xu22], we attempted to investigate the molecular basis of MDR, which may significantly contribute to the prevalence of MDR-TB in study populations. We hypothesized that some dominant M. tuberculosis strains (genotypes) in rural China might possess an increased genetic predisposition to develop MDR and transmission.
Historically, TB disease was considered to be caused by a single strain of M. tuberculosis. However, since molecular genotyping of M. tuberculosis was applied to track the local and global dissemination of the disease, substantial evidence has shown that TB is caused by a variety of M. tuberculosis strains with different biological behaviours in virulence, transmissibility, and competence of developing drug resistance. In a number of previous molecular epidemiological studies, the Beijing family genotype was found most often in outbreak events caused by MDR strains and worldwide dissemination [Reference Glynn14]. The association between this genotype and MDR has been extensively investigated in different geographic regions and populations. We hypothesized that some subgroup strains of the Beijing family may play a significant role in developing MDR, but others may not. Additional genotyping strategies are required to refine the Beijing family and identify the subgroups with increased risk of MDR. A recent study in Shandong Province, China identified a major subgroup of the Beijing family, which was associated with a significantly increased risk of developing RIF resistance [Reference Ma15]. Using a population-based epidemiological study design and two rural Chinese populations with high prevalence of MDR-TB in this study, we continued to investigate the association between this subgroup of the Beijing family and MDR. In this study, we found (1) MDR isolates were remarkably overrepresented (P<0·01) in the isolates with MIRU genotype 223325173533 (44·7%) compared with other Beijing family isolates (13·7%) and non-Beijing family strains (7·4%); (2) MIRU/IS6110 RFLP clustering analysis suggested that this major subgroup of the Beijing family had a significantly increased frequency to be clustered (60·5%) than other Beijing family isolates (21·0%) and non-Beijing family isolates (13·0%); (3) this major subgroup was more likely to cause TB disease in re-treated cases (re-infection, relapse or chronic TB) than other isolates of Beijing family; (4) most clusters from the major group contained INH mono-resistant isolates and MDR-TB simultaneously, which also shared the mutation in katG (S315T) gene, suggesting that isolates with this mutation in the major subgroup (PCR00002) tend to acquire the extra additional mutation related to MDR; (5) CDC strain 210, a widely disseminated Beijing family strain with an average of 21 IS6110 copies, also presents the same MIRU genotype (223325173533) as the PCR00002. CDC strain 210 has been shown to exhibit significantly faster intracellular growth in human macrophages than other Beijing and non-Beijing family isolates [Reference Theus23]. According to data from US National TB Genotyping Service, this genotype (PCR00002) is also one of the most common genotypes in the USA. All these data strongly suggest that this major genotype is an Asian-origin lineage of M. tuberculosis with an increased capacity of transmission, MDR, and survival from anti-TB treatment.
There is evidence that the IS6110 insertion plays an important role in mediating genomic rearrangements and deletions [Reference Fang24]. The IS6110 insertion has also been found to up-regulate downstream genes through an outward-directed promoter in the 3′ end, which may be responsible for the increased capacity of replication, virulence, and transmissibility of CDC strain 210 [Reference Beggs, Eisenach and Cave25, Reference Safi26]. Our data showed that the isolates in the major subgroup (PCR00002) possessed on average more IS6110 insertion elements compared to other isolates in the Beijing family, which could potentially explain the molecular basis underlying these preferential phenotypes of this major subgroup of the Beijing family.
The fitness of M. tuberculosis strains circulating in a community is possibly the driving force perpetuating the TB epidemic. Regarding drug resistance dissemination, it has been suggested that the evolution of drug resistance has a fitness cost resulting in the overall attenuation of the pathogen [Reference Andersson27]. However, this premise might not be true for INH-resistant strains with the Ser315Thr mutation in the katG gene, in which 30–40% of the catalase-peroxidase activity remains [Reference Rouse28]. Several previous studies have also shown that the mutation in position 315 of the katG gene was associated with increased transmissibility, virulence, and drug resistance [Reference Kapetanaki29, Reference van Soolingen30]. In our study, 57·9% of isolates in the major subgroup (PCR00002) carried the katG 315 mutation. This might provide additional evidence that the katG 315 mutation is associated with a selective advantage for M. tuberculosis survival.
The prevalence of this major MIRU genotype within the Beijing family might vary in different areas of China and its association with MDR-TB might vary due to the socio-economic development and performance of DOTS implementation. The predominance and relationship to MDR-TB in the current study could explain its contribution to the epidemic of MDR-TB to some degree. The current study was established in two of China's more than 2000 counties, and the sampled counties were located in the relatively affluent eastern coastal China region. The association between M. tuberculosis and drug resistance might be underestimated compared to that in poorer northern areas, where the situation of MDR-TB might be worse due to poorer access to TB treatment. In addition, MDR-TB has epidemiological significance by causing increased mortality, and longer treatment at a much higher cost. Therefore, MDR-TB and its relevant mutations in hotspot genes were the main concerns of the present study. Additional studies require populations from areas with different DOTS practices and socio-economic development, as well as defining the other drug susceptibilities of the major subgroups and the relevant genetic mutation.
In conclusion, the major subgroup (MIRU type: 223325173533, PCR00002) of the Beijing family is an important genotype of M. tuberculosis, which may have prominent selective advantages, including rapid replication and genetic variability. These unique characteristics could result in increased infectivity, transmissibility and susceptibility to MDR. The prevalence of this major genotype isolates may contribute to the high prevalence of MDR-TB in the rural Chinese population. Its biological characteristics deserve further investigation in animal and human macrophage models. In addition, further genome-level studies will be needed to investigate the molecular specificities of the major subgroup.
ACKNOWLEDGEMENTS
We thank the Houston Tuberculosis Initiative (PI: Edward A. Graviss) for the partial sequencing and genotyping work conducted in this study. We thank the China Scholarship Council (CSC) and Fudan University Graduate School Grant for Innovation for providing support to Yi Hu. We are also grateful to the Katharine H. K. Hsu Foundation and the Vivian L. Smith Foundation for their generous support of Xin Ma for coordinating this US–China collaborative project. This study was approved by the Institutional Review Board of the School of Public Health in Fudan University and supported by grants from National Natural Science Foundation of China (PI: Biao Xu, No. 30771843; PI: Yi Hu, No. 30901223), the China National Key Project for Infectious Disease (grant no. 2009ZX10003-017) and Shanghai Leading Academic Discipline Project (no. B118).
DECLARATION OF INTEREST
None.




