



Introduction: cold denaturation, why it matters
The three-dimensional arrangement of proteins is crucial for their function. In a large number of cases, proteins adopt a defined, albeit not entirely rigid, structure. Understanding the determinants of protein fold stability, that is the ability to retain the three-dimensional structure in the face of environmental insults, is thus inherently essential for a deep understanding of the physical forces that determine protein fold. In in vitro studies, protein unfolding can be caused by different agents, such as for instance heat, pressure, or pH. Each of these agents can reveal specific regions of the energy landscape that determine the folding funnel (Finkelstein et al., Reference Finkelstein, Bogatyreva, Ivankov and Garbuzynskiy2022 and references therein). It is well-accepted that the mechanisms that lead the same protein to unfold under the influence of various denaturing agents are different and, accordingly, the ensembles of conformations of the unfolded states are expected to be different (Pastore & Temussi, Reference Pastore and Temussi2022).
One of the most common denaturing agents used in biophysical studies of unfolding is heat: it is intuitive to expect that most mesophilic proteins unfold at temperatures higher than room temperature because the increase in temperature is known to disrupt many ordered structures. In contrast, lowering the temperature is usually assumed by default to increase stability. This is not true, as protein unfolding also occurs at low temperatures. Hints of the possible destabilization of proteins at low temperatures date back to the 30s. The identification of several early examples of proteins undergoing cold denaturation is quoted in a well-known review by the Russian Petr Privalov, one of the fathers of modern protein thermodynamics (Privalov, Reference Privalov1990). For instance, Hopkins reported that the rate of denaturation of ovalbumin by urea is higher at 0 °C than at 23 °C (Hopkins, Reference Hopkins1930). This observation was soon backed up by several similar studies (Jacobsen and Christensen, Reference Jacobsen and Christensen1948; Simpson and Kauzmann, Reference Simpson and Kauzmann1953; Christensen, Reference Christensen1952; Nojima et al., Reference Nojima, Ikai, Oshima and Noda1977), but it took many more years before the existence of cold denaturation became fully accepted by the scientific community.
Despite its importance, it is clear that cold denaturation has received relatively little interest. This is mostly because its occurrence, for most mesophilic proteins, is expected at temperatures much lower than zero degrees. The most common way to observe cold denaturation has been to raise the temperature at which cold denaturation occurs, by destabilizing the protein through ad hoc mutations and/or by adding denaturants. For example, Antonino et al. (Reference Antonino, Kautz, Nakano, Fox and Fink1991) observed cold denaturation in a mutant of staphylococcal nuclease, named NCA S28G, containing the point mutation Ser28->Gly. Hatley and Franks (Reference Hatley and Franks1989) were able to observe cold denaturation of lactate dehydrogenase by adding large amounts of methanol. Several groups used guanidine hydrochloride to destabilize proteins. With this denaturant, Azuaga et al. (Reference Azuaga, Galisteo, Mayorga, Cortijo and Mateo1992) observed cold denaturation of beta-lactoglobulin B, and Agashe and Udgaonkar (Reference Agashe and Udgaonkar1995) measured cold denaturation of barstar. The drawback of this strategy, based on the artificial denaturation of proteins, is that denaturation occurs under conditions very different from physiological conditions.
In 2007, we identified a protein, Yfh1, whose cold denaturation occurs at temperatures close to room temperature and under (quasi) physiological conditions (Pastore, Martin, et al., Reference Pastore, Martin, Politou, Kondapalli, Stemmler and Temussi2007), having the two observable melting temperatures around 5 and 35 °C when studied in solutions of low ionic strength. Although these conditions are harsh on proteins which are usually exposed to appreciable ionic strength, low salt is in no way comparable or as disruptive as the addition of strong denaturants. The possibility of measuring both heat and cold denaturation allows the direct measurement of the ∆Cp and hence the determination of the temperature dependence of the difference in free energy between the folded and unfolded states, known as the stability curve (Becktel & Schellman, Reference Becktel and Schellman1987). In turn, the easy determination of the stability curve provides a better way of judging the influence of external factors on protein stability, much more significant than the single heat denaturation temperature or the parameter m, which is related to the increase in the degree of exposure of the protein upon chemical denaturation (Pace, Reference Pace1986). Yfh1 is thus uniquely suited for making an in-depth characterization of the factors influencing the stability of a natural full-length protein, as a function of temperature and much more. Using Yfh1, the studies of unfolding caused by different agents on the same protein opened up the possibility of clarifying the nature of the different unfolded states and shedding light on the mechanisms of unfolding.
In the present review, we wish to summarize all of what has been learned about protein stability thanks to the unique properties of Yfh1 and describe in detail the open questions that Yfh1 has helped to address, together with the biophysical aspects that these studies clarified on the nature of protein stability.
Cold denaturation in a nutshell
The occurrence of the cold denaturation transition is a direct consequence, from a thermodynamic point of view, of the protein stability curve that is described by the so-called modified Gibbs–Helmholtz equation:
 $$ \Delta G=\Delta {H}_m\left[1-\frac{T}{T_m}\right]+\Delta {C}_p\left\{\left(T-{T}_m\right)- Tln\left[\frac{T}{T_m}\right]\right\} $$
$$ \Delta G=\Delta {H}_m\left[1-\frac{T}{T_m}\right]+\Delta {C}_p\left\{\left(T-{T}_m\right)- Tln\left[\frac{T}{T_m}\right]\right\} $$
where Tm is the temperature at the midpoint of the unfolding transition, and ∆Hm is the unfolding enthalpy change at Tm. When both high and low temperature unfolding temperatures are accessible, it becomes possible to determine the heat capacity difference between the two states and thus calculate the full stability curve of the protein (Pastore et al., Reference Pastore, Martin and Temussi2019).
Becktel and Schellman (Reference Becktel and Schellman1987) have shown that the free energy difference between the unfolded and the native state (∆G), when plotted versus temperature, is bell-shaped and convex, with a maximum at a temperature of maximal stability that is close to room temperature (TS) for proteins from mesophilic organisms. Therefore, destabilization of the native state occurs as the temperature varies from room temperature in either direction, when the curve crosses the two zero points of ∆G. Analysis of the temperatures of unfolding of proteins both at high and low temperatures gives a better quantitative measure of stability and allows a more accurate identification of the effects of different denaturation agents: addition of, say, a co-solvent or introduction of a mutation may affect differently the cold and heat denaturation temperatures but also lower the Ts with an overall reduction of stability and cooperativity of the unfolding process. In 2017, Temussi and coworkers proposed the use of a single parameter to describe thermal stability considering the area of the stability curve between the two unfolding temperatures (Alfano et al., Reference Alfano, Sanfelice, Martin, Pastore and Temussi2017). To compare areas, it is possible to calculate the integral of the stability curve described by Eq. (1) and calculate its value between T c and T m.
The integral is given by:
 $$ I={\displaystyle \begin{array}{l}-\left(\Delta {H}_m\frac{T^2}{2{T}_m}\right)+\Delta {H}_mT-\frac{1}{2}\Delta {C}_p{T}^2\;\ln \left(\frac{T}{T_m}\right)-\Delta {C}_p{T}_mT\\ {}+\hskip2px \frac{3}{4}\Delta {C}_p{T}^2\end{array}} $$
$$ I={\displaystyle \begin{array}{l}-\left(\Delta {H}_m\frac{T^2}{2{T}_m}\right)+\Delta {H}_mT-\frac{1}{2}\Delta {C}_p{T}^2\;\ln \left(\frac{T}{T_m}\right)-\Delta {C}_p{T}_mT\\ {}+\hskip2px \frac{3}{4}\Delta {C}_p{T}^2\end{array}} $$
Although empirical and without a direct physical meaning, this parameter is an assessment of the global thermodynamic stability within the entire temperature range of protein stability, rather than at a single temperature.
The ID card of Yfh1
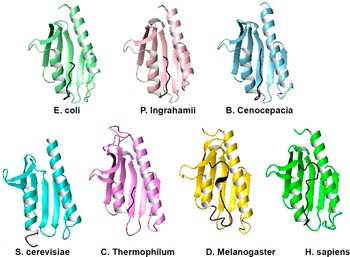
Yfh1 is the yeast orthologue of frataxin and a member of a protein family highly conserved from bacteria to primates and involved in crucial metabolic processes, being essential for the biosynthesis of sulphur–iron clusters (Want et al., Reference Want and D’Autréaux2024). Human frataxin is associated with the recessive neurodegenerative disease Friedreich’s ataxia (Pastore & Puccio, Reference Pastore and Puccio2013). This disease is caused by a usually homozygous trinucleotide expansion in the non-coding region of the FXR1 gene which leads to reduced expression levels of the protein. A minor percentage of Friedreich’s ataxia patients (ca., 2%) is heterozygous with the expansion on one allele and missense mutations or truncations on the other. Frataxins are in fact small proteins that are so essential to metabolism that their reduced expression causes oxidative stress and hampers the formation of iron–sulphur clusters, heme synthesis, and mitochondrial function. In eukaryotes, they are nuclear-encoded, translated into the cytoplasm, and then imported into mitochondria, where they are matured. Accordingly, sequence alignment of the family has shown that frataxins have a rather variable N-terminus and a highly conserved globular C-terminal domain (Figure 1). The N-terminus is absent in prokaryotes and is a linker with the signal peptide that is eventually cleaved off after maturation. When present, this region is disordered and highly mobile as demonstrated for Yfh1 (Popovic et al., Reference Popovic, Sanfelice, Pastore, Prischi, Temussi and Pastore2015).

Figure 1. Multiple alignments of the sequences of organisms for which the 3D structure is now available. The first three sequences correspond to prokaryotic proteins, and the last four come from eukaryotes. The alignment shows clearly how the prokaryotes do not have a signal peptide that has been truncated during processing leaving in the mature protein an N-terminal tail that is absent in prokaryotes. Note that we are aligning the eukaryotic sequences before maturation which thus contain the signal peptides. The beginning of the mature forms of the human and yeast proteins are indicated with red and green arrows, respectively.
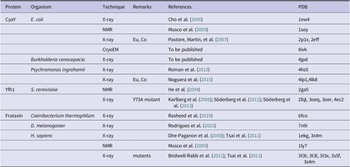
The structures of frataxin orthologues from seven different species are currently available (Table 1). Three of them are from bacteria and four more are from eukaryotic organisms. The fold of the conserved domain is compact and globular with no cavities or grooves and contains a β-sheet of 5–7 strands packed against two helices that shield the hydrophobic face of the sheet together with a C-terminal tail (Figure 2). It was noted early on that ‘This [C-terminus] must be crucial for the stability of the fold as it allows the formation of extensive interactions between the hydrophobic residues that are conserved in the frataxin family’ (Musco et al., Reference Musco, Stier, Kolmerer, Adinolfi, Martin, Frenkiel, Gibson and Pastore2000). It was in fact observed that different frataxin orthologues differ strongly in the length of the C-terminus. When present, this secondary structural element inserts in between the two terminal helices, thereby protecting the hydrophobic core.
Table 1. List of the available structures of frataxin orthologs with their PDB accession names and the relative references
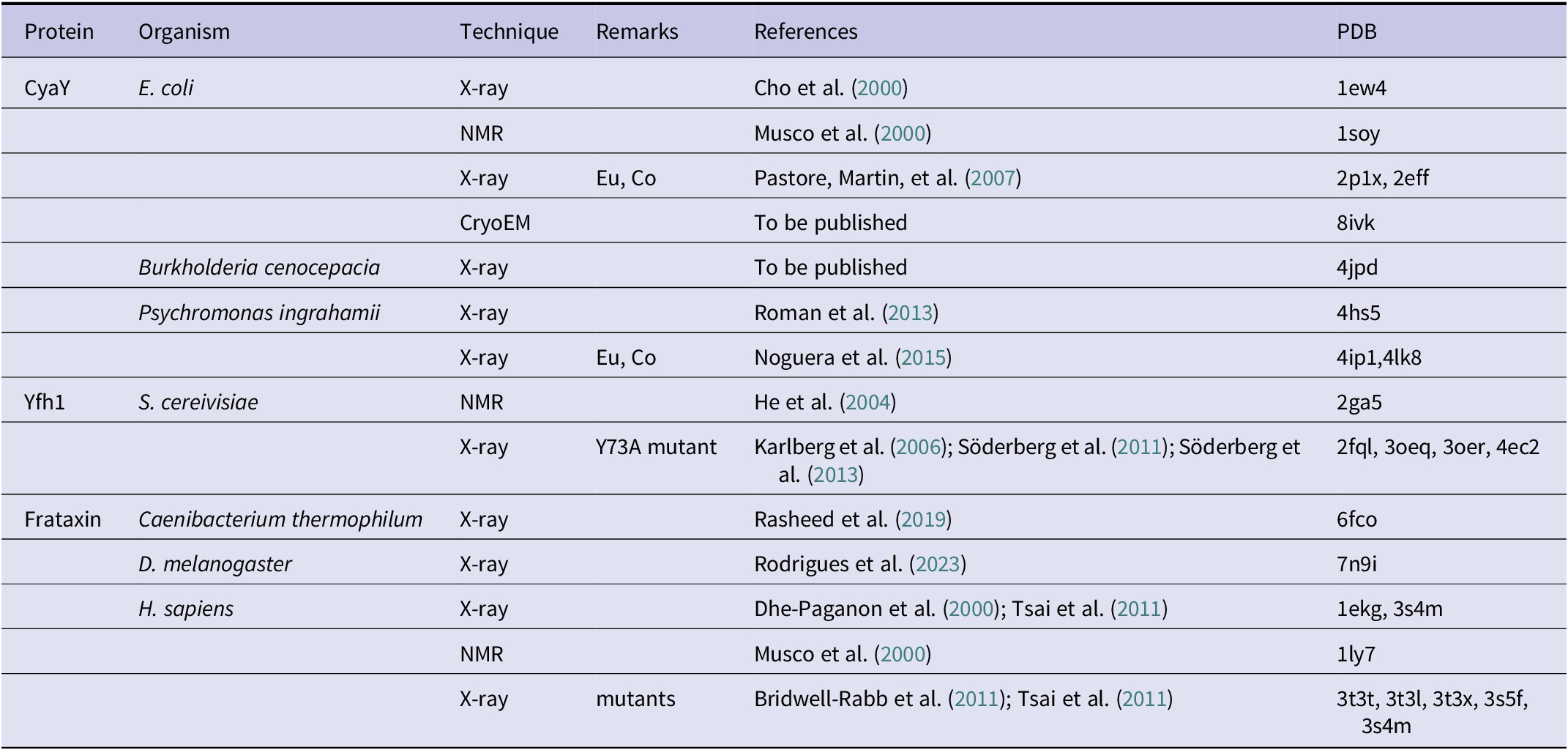

Figure 2. Ribbon representation of a representative selection of the available structures of frataxin orthologues from different species. Many more structures are available, especially for the human and yeast proteins, crystallized under different conditions and bound to various cations and/or in different molecular complexes but here we show only representative structures for each of the species. When present, the C-terminal extension is indicated in black. The comparison makes clear how this extension inserts between the two helices and protects the hydrophobic core. The absence of the extension leaves the core much more exposed and vulnerable so that the entropic motions induced by high temperature promptly unfold the protein. The corresponding PDB codes are (from left to right and from top to bottom): 1ew4, 4hs5, 4jpd, 2ga5, 6fco, 7n9i, and 1ekg.
Another hallmark of the family is that its members are acidic proteins with isoelectric points around 4.9 (Adinolfi et al., Reference Adinolfi, Nair, Politou, Bayer, Martin, Temussi and Pastore2004). An even superficial look at the available structures shows that the negative charge distribution is uneven, with a high concentration on the first helix and the first two β-strands of the sheet (Figure 3A). Some of the negatively charged residues are semi-conserved and they all cluster on one side of the protein surface, encompassing the solvent-exposed face of the first helix and the beginning of the β-sheet (Musco et al., Reference Musco, Stier, Kolmerer, Adinolfi, Martin, Frenkiel, Gibson and Pastore2000). Remarkably, this is the same surface containing clinically important mutations (Musco et al., Reference Musco, Stier, Kolmerer, Adinolfi, Martin, Frenkiel, Gibson and Pastore2000). Given that the protein is monomeric in solution, the conservation of exposed residues could only be explained by the formation of a functional surface. In hindsight, we now know that this region is indeed needed to bind to iron and interact with a positively charged patch on the surface of the main known frataxin partner, the desulfurase enzyme that is central in iron–sulphur cluster formation (Prischi et al., Reference Prischi, Konarev, Iannuzzi, Pastore, Adinolfi, Martin, Svergun and Pastore2010). The interaction is mainly electrostatic and thus strict conservation of the residues involved is not required.

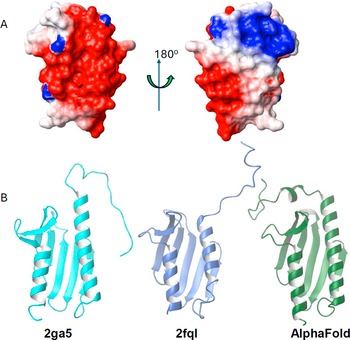
Figure 3. Charge distribution and comparison of the available structures of Yfh1. (a) Electrostatic potential on the surface of Yfh1 (2ga5) shown in two different views rotated by 180° around the y axis. The structure on the left has the same orientation in Figure 1. (b) Left: NMR structure; Middle: representative structure of a mutant of Yfh1 designed to enhance the ability of the protein to aggregate and form big complexes; Right: an AlphaFold model.
It should be noted that there are two structures for yeast Yfh1 in PDB: 2ga5 is an NMR structure that has some geometric distortions (Vilanova et al., Reference Vilanova, Sanfelice, Martorell, Temussi and Pastore2014), and a series of crystallographic structures at about 3 Å resolution of a mutant of the conserved Tyr73 to alanine (Söderberg et al., Reference Söderberg, Shkumatov, Rajan, Gakh, Svergun, Isaya and Al-Karadaghi2011). This mutation enhances the tendency of the protein to form large aggregates in the presence of iron and affects the structure of the N-terminal helix that hosts Tyr73 shortening it by the first two helical turns (L70-E76) that are instead present in the NMR structure (Figure 3B). The last two C-terminal residues (S172, Q173) are also not detectable in the crystal structure. A model with a high degree of confidence obtained with the AlphaFold version 2.2 software superposes with the crystal structure with a 1.1 Å rmsd in the globular region (residues 75–172) and with the NMR structure with a 2.2 Å rmsd. This model is probably, for the time being, more representative of the structure of Yfh1 than either of the NMR and crystal structures.
Comparison of the frataxin structures and their high-temperature stabilities
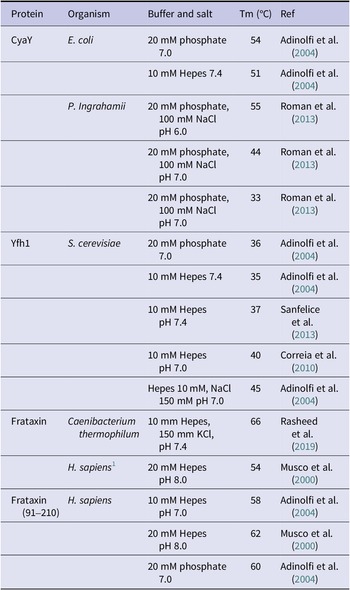
Studies exploring the factors influencing this interesting family of proteins started shortly after the first structural determinations (Dhe-Paganon et al., Reference Dhe-Paganon, Shigeta, Chi, Ristow and Shoelson2000; Musco et al., Reference Musco, Stier, Kolmerer, Adinolfi, Martin, Frenkiel, Gibson and Pastore2000). Adinolfi et al. (Reference Adinolfi, Nair, Politou, Bayer, Martin, Temussi and Pastore2004) carried out a comparative structural study of frataxin orthologs from three organisms of different evolutionary history: Escherichia coli (CyaY), Saccharomyces cerevisiae (Yfh1), and Homo sapiens (frataxin). The authors showed that the proteins have the same fold but also, when studied under the same conditions, remarkably different melting temperatures: the heat-induced melting temperatures at low salt of the yeast, bacterial, and human proteins go from 35, 55, and up to 65 °C respectively, Yfh1 is thus by far the least stable of these proteins and seemed to contain in solution an appreciable amount of a disordered species in equilibrium with the folded species.
In trying to understand the reasons for such different stabilities, it was noticed that the melting points of the three model systems correlate with the length of the C-terminus. From the multiple alignment of the family, it is clear that the C-terminus of Yfh1 is shorter compared to all other orthologues (e.g., it is 3 residues shorter than the E. coli orthologue and 12 residues shorter than the human protein) (Adinolfi et al., Reference Adinolfi, Nair, Politou, Bayer, Martin, Temussi and Pastore2004). It was thus hypothesised that the results could be explained by assuming that the hydrophobic core of Yfh1 is more easily accessible because the C-terminus that helps to protect the core is absent. To prove this hypothesis, it was shown that shortening the human protein by producing a truncated mutant caused a major destabilization which drastically reduced the Tm of the protein, whereas lengthening the yeast protein led to stabilization of about 10 °C in ΔTm (Adinolfi et al., Reference Adinolfi, Nair, Politou, Bayer, Martin, Temussi and Pastore2004). These conclusions should not be understood as ‘the length of the C-terminus of frataxins is the cause of their stability’, since protein stability is the resultant of several different attractive and repulsive interactions. All it means is that the length of the frataxin C-terminus is a factor that strongly influences stability. We now have the structures and melting points of other frataxins from different organisms and the original hypothesis still roughly holds (Table 2).
Table 2. Comparison of the available heat melting temperatures of frataxins from different organisms

Note: The table is meant to give an indication of the variability of the stability among the different proteins but also of the sensitivity of each protein to the environmental conditions. When possible, we have tried to report similar conditions.
1 This refers to the full-length mature human frataxin that is made of the conserved domain found in all frataxin and an unstructured N-terminal tail of 28 residues. As it can be appreciated by comparison of the stability of this protein with that of the isolated domain (Frataxin(91–210)), the unstructured tail slightly destabilizes the protein.
The marginal stability of Yfh1 and cold denaturation
At first sight, it would be natural to think that it ought to be difficult to measure the full curve of stability of a marginally stable protein, but this is not true (Figure 4). A stable protein, typically with a free energy difference between unfolded and folded states around 10 kcal/mole (dashed curve), has a temperature of cold denaturation far below the freezing point of water and is therefore inaccessible, but if one lowers the maximum of the curve to about 1 kcal/mole (solid curve) the cold denaturation point becomes close to the freezing point of water. Accordingly, in 2007, it was serendipitously discovered that the cold denaturation midpoint of Yfh1 is not, as in many mesophilic proteins well below the freezing point of water and can instead be observed at temperatures close to but above 0 °C (Pastore, Martin, et al., Reference Pastore, Martin, Politou, Kondapalli, Stemmler and Temussi2007). It is thus possible to observe cold denaturation of Yfh1 precisely thanks to its marginal stability. This observation opened an entirely different new world.

Figure 4. Accessibility of cold denaturation. The dashed curve, with a ΔGUF of approximately 10 kcal/mole at maximum stability has a cold denaturation temperature close to 230 K. Translation to a ΔGUF of approximately 1 kcal/mole (solid curve) raises the cold denaturation point very close to 273 K.
Analysis of the thermal unfolding curves of Yfh1 recorded by far-UV circular dichroism indicated melting points for the heat and cold transitions around 35 and 5 °C respectively. Calculation of ∆H at the high-temperature melting point gave a value of about 35 kcal/mol. Inserting this value in the Gibbs-Helmholtz equation gave an estimated ∆Cp of 1.7 kcal (K mol)−1 and a free energy of unfolding at 20 °C between 0.5 and 1 kcal/mole, depending on the exact solution conditions, suggesting that Yfh1 is only 70%–87% folded at 20 °C. The unfolding of Yfh1 is however highly cooperative as demonstrated in several papers (Adinolfi et al., Reference Adinolfi, Nair, Politou, Bayer, Martin, Temussi and Pastore2004; Sanfelice et al., Reference Sanfelice, Politou, Martin, De Los Rios, Temussi and Pastore2013, Reference Sanfelice, Morandi, Pastore, Niccolai and Temussi2015) and fully reversible under all the unfolding conditions explored without noticeable aggregation.
It should be noticed though that, despite the low stability of Yfh1, we are not in the presence of a molten globule, which is a protein that adopts a secondary but not a tertiary structure (Ptitsyn, Reference Ptitsyn1995). This could be clearly excluded from the NMR spectrum of Yfh1: the 15N 2D HSQC spectrum at room temperature is typical of a protein with a well-defined structure as can be appreciated by the exquisite spread of the resonances in both dimensions (Figure 5). Careful analysis and assignment of the spectrum showed however that we are in the presence of two completely distinct populations: one predominant at room temperature corresponding to the folded species, and a second one, less represented, corresponding to the unfolded species. They are in slow equilibrium so that, by increasing the temperature, the folded species weakens and eventually disappears while the unfolded species becomes predominant. We thus concluded that Yfh1 is a marginally stable protein, rather than an intrinsically disordered protein or a molten globule state (Pastore et al., Reference Pastore, Martin and Temussi2019).
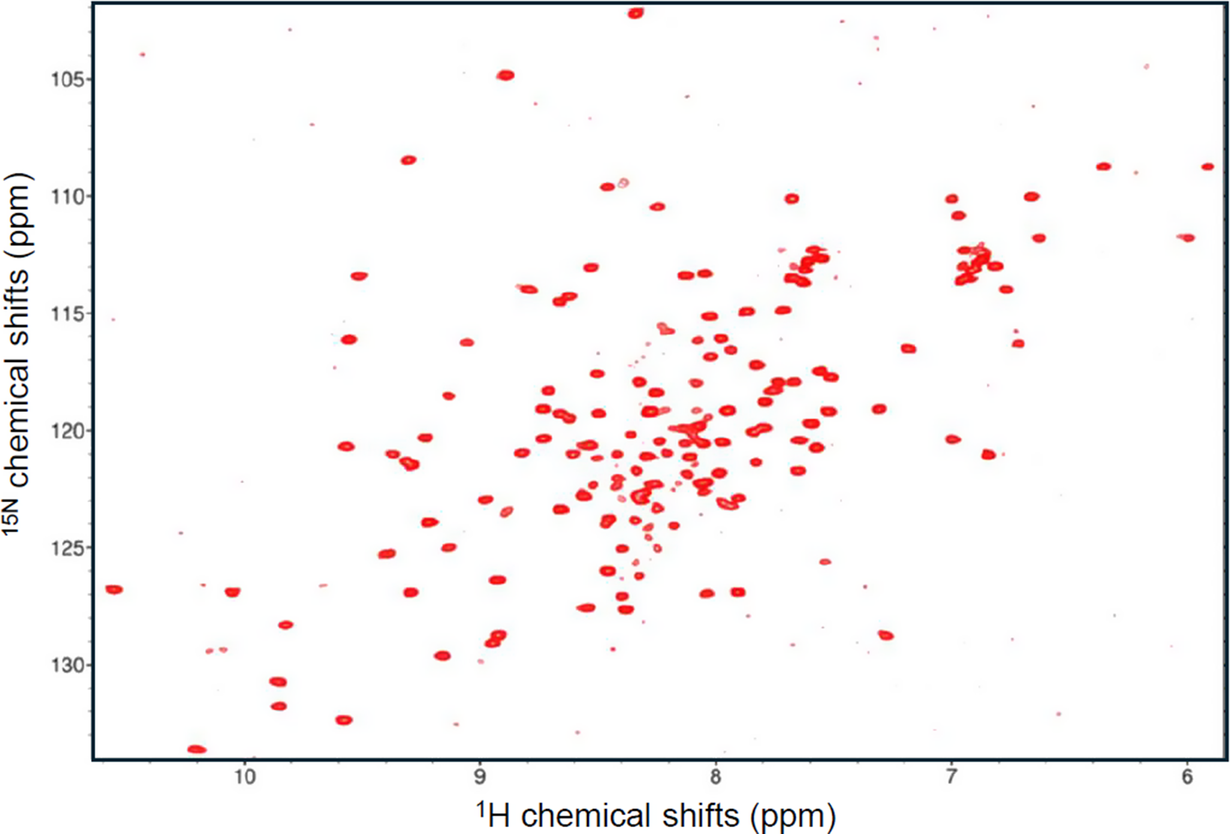
Figure 5. HSQC spectrum of Yfh1 at 20 °C and room pressure recorded at 600 MHz. The protein is in 20 mM Hepes at pH 7.0. The weak peaks especially around 8.2 and 124 ppm but also distributed throughout the spectrum correspond to the unfolded form. As proven by Vilanova et al., Reference Vilanova, Sanfelice, Martorell, Temussi and Pastore2014, they disappear upon the addition of 100 mM NaCl.
Searching for the causes of cold denaturation of Yfh1
The fascinating properties mentioned above prompted the search for factors that could explain this unusual behaviour. In 2010, Correia et al. reported the characterisation of eight Yfh1 variants affecting residues in the α1/β1 acidic ridge identified in Musco et al. (Reference Musco, Stier, Kolmerer, Adinolfi, Martin, Frenkiel, Gibson and Pastore2000) and the conserved β-sheet surface (Correia et al., Reference Correia, Wang, Craig and Gomes2010). The authors demonstrated that these mutations did not affect the iron binding capacity but significantly decreased the stability in agreement with the hypothesis of direct competition between function and stability (Masino et al., Reference Masino, Nicastro, Calder, Vendruscolo and Pastore2011; Pastore & Temussi, Reference Pastore and Temussi2012). Mutation of up to five negatively charged residues resulted in a thermal stabilization of ~24 °C. As a negative control, mutation of conserved residues on the β-sheet residues had only a modest impact on the protein stability. While these results were interesting in view of an intrinsic competition between physiological function and stability which was the focus of the work, surprisingly, the authors made no attempt to check whether their mutations had any influence on the cold denaturation transition: the measurements were all carried out at 50 mM salt concentrations and in the range 10 to 90 °C. Under these conditions, cold denaturation would not be detectable.
In 2013 and a more recent study (González-Lebrero et al., Reference González-Lebrero, Defelipe, Modenutti, Roitberg, Batastini, Noguera, Santos and Roman2019; Roman et al., Reference Roman, Faraj, Cousido-Siah, Mitschler, Podjarny and Santos2013), Santos and coworkers studied the frataxin ortholog from Psychromonas ingrahamii, a psychrophilic microorganism that proliferates at low temperatures (cold adaptation), where natural selection overcomes the problems of cold denaturation, slow protein folding dynamics, and reduced enzyme activities. The authors studied in detail the pH dependence of the heat denaturation of the protein and its behaviour under chemical denaturation, but, also in this case, all the studies were carried out in the presence of salt, and temperature scanning experiments never covered temperatures below 10 °C, preventing a priori the possibility of observing the region of the spectra most interesting for the detection of cold denaturation.
A different study was conducted soon after to check the effects of salt on cold denaturation (Sanfelice et al., Reference Sanfelice, Politou, Martin, De Los Rios, Temussi and Pastore2013). It had in fact already been noticed that cold denaturation became undetectable if even small concentrations of monovalent salts were added. It was thus hypothesized that repulsion among spatially close negative charges on the protein surface could facilitate the access of water molecules into the hydrophobic core, according to Privalov’s theories (Privalov, Reference Privalov1990). Yfh1, but not other orthologues, contains four negatively charged residues on the first and second strands of the sheet (Sanfelice et al., Reference Sanfelice, Politou, Martin, De Los Rios, Temussi and Pastore2013) (Figure 6). It was argued that this quadrilateral of negative charges could cause significant electrostatic frustration which could lead to cold denaturation under conditions in which hydrophobic forces are weaker. To prove this hypothesis, the same authors introduced in a following study single, double, and triple mutations and observed that even mutation of only one of these residues to a neutral hydrophilic group leads to the shift of cold denaturation temperatures to below the water freezing point while not altering significantly the high-temperature melting point (Sanfelice et al., Reference Sanfelice, Morandi, Pastore, Niccolai and Temussi2015). It is noteworthy that some of the mutated residues were the same as those mutated in Correia et al. (Reference Correia, Wang, Craig and Gomes2010).

Figure 6. Comparison of the charge distributions among some members of the frataxin family showing the residues that induce electrostatic frustration in Yfh1. Light green: E. coli (1ew4); Pink: P. Ingrahamii (4hs5); Cyan: Saccharomyces cerevisiae (2ga5); Bright green: H. sapiens (1ekg). The sidechains of glutamates and aspartates are shown in red, and those of lysines and arginines are in blue. The residues corresponding to the quadrilateral that destabilizes Yfh1 are shown with a thicker bond radius. Notice that the distribution of charges in P. Ingrahamii and S. cerevisiae is very similar. The only potentially stabilizing element is lysine 40. It can thus be predicted that, if properly destabilized C-terminally, the frataxin orthologue from Psychromonas ingrahamii is expected to give cold denaturation either as it is or by neutralizing lysine 40.
In support of this hypothesis, it was then reasoned that, if one could manage to induce detectable cold denaturation in an otherwise stable protein, this would prove conclusively that the rules that governed the cold denaturation of Yfh1 were understood. Indeed, it was possible to convert CyaY, a stable bacterial orthologue of Yfh1, to a marginally stable protein that undergoes detectable cold denaturation by inducing electrostatic frustration in the bacterial protein through a few mutations corresponding to those destabilizing Yfh1 (Bitonti et al., Reference Bitonti, Puglisi, Meli, Martin, Colombo, Temussi and Pastore2022). It is, however, important to stress that, to observe cold denaturation, it was first necessary to C-terminally truncate the protein to reduce its high-temperature melting point from 50 to 35 °C, which is close to what is observed for Yfh1. Care was also taken to mutate nearby positive charges that could compensate for the negatively charged repulsion. A posteriori, given the evidence collected, it is easy to predict that, if properly destabilised C-terminally, the protein from P. ingrahamii has almost all the requisites necessary to give detectable cold denaturation and would then be possibly only one single mutation away from cold denaturation (Figure 6). Hopefully, this prediction will soon be tested because it could suggest some interesting hypotheses on evolutionary strategies for cold resistance.
Cold denaturation: environment or molecularity?
These results are interesting in view of understanding the causes of the cold denaturation phenomenon. Its occurrence was explained on the basis of thermodynamic considerations by Privalov (Reference Privalov1990), who proposed that low temperature favours the preferential solvation of nonpolar side chains, a phenomenon that does not occur at high temperatures. He argued that the free energy of hydration is negative with an absolute value that increases when the temperature goes down. Thus, at a sufficiently low temperature, the non-polar groups of the hydrophobic core are forced to be exposed to water causing the unfolding of an otherwise compact protein (Privalov, Reference Privalov1990).
The mechanism postulated by Privalov (Reference Privalov1990), although entirely convincing on its thermodynamic basis, has often been the object of heated controversy by researchers attributing cold denaturation to a series of physical causes, which were mainly linked to properties of water (see for instance Kauzmann, Reference Kauzmann1959; Graziano et al., Reference Graziano, Catanzano, Riccio and Barone1997). After the discovery of the behaviour of Yfh1, there was a substantial increase in papers dealing with the origin of cold denaturation. Graziano (Reference Graziano2010, Reference Graziano2014) reasoned that the water-accessible surface area (WASA) has a great influence on the very different conformations that a polypeptide chain can adopt. According to this author, there would be a temperature where the destabilizing contribution of the polypeptide chain conformational entropy matches exactly the stabilizing contribution of the water configurational/translational entropy, leading to cold denaturation. Yoshidome and Kinoshita (Reference Yoshidome and Kinoshita2012) attributed cold denaturation to the weakening of hydrophobicity at low temperatures, according to several experimental studies. Gulevsky and Relina (Reference Gulevsky and Relina2013), by reviewing many cases of cold denaturation, concluded that the disorganisation of supramolecular assemblies under the influence of low temperature is one of the key factors contributing to cell cold-induced injuries. Bianco and Franzese (Reference Bianco and Franzese2015) showed, using Monte Carlo simulations, that it is sufficient to take into account how water at the protein interface changes its hydrogen bond properties and its density fluctuations for predicting protein stability regions with elliptic shapes in the temperature–pressure plane, consistent with previous theories. Recently, Seelig and Seelig (Reference Seelig and Seelig2023) gave an original interpretation of thermal protein unfolding, including cold denaturation. They demonstrated that the temperature profiles of enthalpy ΔH(T), entropy ΔS(T), and free energy ΔG(T) can be obtained directly by a numerical integration of the heat capacity profile Cp(T) that can be obtained by differential scanning calorimetry (DSC), a technique that offers the unique possibility to assess these parameters without resorting to a model. They introduced three new models, an empirical two-state model, a statistical–mechanical two-state model, and a cooperative statistical–mechanical multistate model. The two-state models yielded good fits for the enthalpy, entropy, and free energy of the unfolding of small proteins. The cooperative statistical–mechanical multistate model yielded an even better fit, even for the unfolding of larger proteins such as antibodies.
However, in our opinion, in most studies on cold denaturation, too little attention has been paid to molecular aspects concerning the specific nature of the protein under study, focusing almost exclusively on the bulk properties of the medium. The experimental data reported by Sanfelice et al. (Reference Sanfelice, Morandi, Pastore, Niccolai and Temussi2015) and the identification of specific destabilizing effects emphasized instead the importance of molecular mechanisms and the specific contribution of each protein in cold denaturation studies, entirely shifting the paradigm from ‘solvent-properties-only’ to ‘protein-properties’.
Yfh1 as a unique tool in protein studies: the effect of alcohols
A consequence of the unusual possibility to measure the stability curve of a protein yields a measure of protein stability more informative than many other parameters, allowing the investigation of the influence of many different perturbing agents. A class of substances commonly used to access cold denaturation are, in addition to chaotropic agents, alcohols, which are known to destabilize proteins and thus increase the temperature of the cold denaturation transition when added to buffer solutions at fairly high concentrations. The general belief is/was that the observation of cold denaturation above zero centigrade in alcohols is possible because hydroalcoholic solutions have freezing temperatures lower than zero degrees. However, it was completely ignored that the very presence of alcohol might change protein stability because the chemical would inevitably alter the composition of the aqueous solution. Martin et al. (Reference Martin, Esposito, De Los Rios, Pastore and Temussi2008) studied the stability of Yfh1 in hydroalcoholic solutions in the presence of three alcohols, that is, methanol, ethanol, and trifluoroethanol. These alcohols were used at concentrations in the range of 0 to 15%, The authors discovered that at these low concentrations, alcohols extend the temperature range in which proteins are stable rather than having a destabilizing effect. The authors determined that the extended temperature range stems from a modest increase in the high temperature of unfolding accompanied by a marked decrease in the cold denaturation transition, thus demonstrating that the effects on the two transitions may be asymmetric. These experimental findings were interpreted on the basis of a direct interaction of the folded conformation with alcohols and of an indirect interference of alcohols with the hydration of the unfolded conformation at low temperatures, completely changing our perception of the effect of alcohols effects on proteins.
Cold denaturation as a way to explore molecular crowding
The ‘tool Yfh1’ was also deemed to be ideal for studies of the influence of crowding on protein stability and in particular, it offered a chance for a comparison of the effects of crowding and confinement on the same protein. The cell interior is often described as being both crowded and confining (Alfano et al., Reference Alfano, Fichou, Huber, Weiss, Spruijt, Ebbinghaus, De Luca, Morando, Vetri, Temussi and Pastore2024). However, the two terms indicate two distinct conditions. Crowding describes a dynamic situation in which available volume is excluded whereas confinement refers to a static condition, characterized by both shape and volume restrictions (Ellis, Reference Ellis2001; Zhou, Reference Zhou2004; Zhou et al., Reference Zhou, Rivas and Minton2008). Based on entropic considerations, it is generally believed that crowding and confinement increase the stability of proteins because the more compact conformations of the folded native states are favoured with respect to the bulkier and more disordered unfolded states. Sanfelice et al. (Reference Sanfelice, Politou, Martin, De Los Rios, Temussi and Pastore2013) attempted a comparison of the effects of crowding and confinement in a rigorous way by studying them using Yfh1. The authors used CD spectroscopy for the comparison between stability in a buffer and a crowded environment, and NMR spectroscopy for the comparison between stability in a buffer and a confining environment. The thermodynamic parameters found by Sanfelice et al. (Reference Sanfelice, Politou, Martin, De Los Rios, Temussi and Pastore2013) showed that crowding has a strong influence on stability: in crowded conditions, there is indeed an increase of the heat unfolding temperature (Tm) but also an increase in enthalpy and a decrease in heat capacity. Furthermore, their results showed that the effects of crowding can be nonlinear in the crowding agent concentration. The authors concluded that, under the conditions of their study, both crowding and confinement influenced the stability of Yfh1. A more quantitative study on the effects of crowding was performed on Yfh1 by Alfano et al. (Reference Alfano, Sanfelice, Martin, Pastore and Temussi2017). At the time this article was published, there were several debates concerning the importance of crowding, both in vitro and in vivo. A survey of the literature suggested that the effects of excluded volume on protein stability are modest. Most of all, according to some authors the predominant influence of crowders is the enthalpic effect of weak interactions rather than entropic effects (Smith et al., Reference Smith, Zhang, Pielak and Li2015). Miklos et al. (Reference Miklos, Sarkar, Wang and Pielak2011), for instance, found that protein crowders can be mildly destabilizing. The competition between entropic and enthalpic effects yields tuneable stability, a situation that is crucial for understanding the role of proteins in living systems. Other authors hypothesized that the volume variations associated with the transition from folded to unfolded species were overestimated (Politou & Temussi, Reference Politou and Temussi2015).
There were, however, various difficulties in discriminating between these possibilities on the effect of crowding on protein stability, mainly because of the uncertainty over the choice of the protein and of the most suitable crowder and its concentration.
The answer to the choice of protein was obvious considering the properties of Yfh1. This protein provided a perfect tool for two reasons: it is marginally stable and thus even small effects on its stability can be easily detected but, even more importantly, it allows the easy determination of the full stability curve. Alfano et al. (Reference Alfano, Sanfelice, Martin, Pastore and Temussi2017) studied the influence, on the stability of Yfh1, of different macromolecular synthetic crowders at concentrations ranging between 5% and 20% w/v taking extra care to ascertain that they did not interact with Yfh1 before assessing their role as crowders. This comparison highlighted the importance of the relative dimensions of the protein under study and of the crowders. Crowders whose molecular weight was closer to that of the protein under study appeared to be more effective than larger crowders in stabilizing the protein. Altogether, these authors demonstrated that there is a clear entropic effect of crowders on protein stability.
NMR studies of protein unfolding
The use of Yfh1 was also helpful in clarifying the use of NMR in studies of unfolding.
NMR has a unique advantage, compared to many other biophysical techniques, in providing residue-specific information on the behaviour of selected residues. However, one might wonder whether all residues are equally suited for the purpose. Protein unfolding can, for instance, be monitored by one-dimensional (1D) NMR by following at different temperatures the resonances of residues spatially close to aromatic rings (ring-current shifted resonances) (Pastore, Martin, et al., Reference Pastore, Martin, Politou, Kondapalli, Stemmler and Temussi2007; Szyperski et al., Reference Szyperski, Mills, Perl and Balbach2006). These residues are, by definition, excellent reporters since they are buried in the hydrophobic core. More difficult is the application of multi-dimensional NMR that provides information on all residues (provided that they are not overlapping). Puglisi et al. (Reference Puglisi, Brylski, Alfano, Martin, Pastore and Temussi2020) analysed this problem and realized that, when looking at individual residues, the position of the reporter group in the protein structure may reflect cooperative unfolding only if the atom is well inside the hydrophobic core. Otherwise, if the chemical group is positioned on external structural elements, volume changes reflect peripheral motions.
To circumvent the problem, the authors first selected the NH resonances of an HSQC from residues as close as possible to the hydrophobic core on the basis of the solvent-accessible surface area (SASA) (Puglisi et al., Reference Puglisi, Brylski, Alfano, Martin, Pastore and Temussi2020). However, they realised that the approach could be misleading because these quantities are calculated for the whole residue rather than for the specific indole group. As an alternative, the authors defined a quantity RAD = (D × RA × 100), where D is the depth of a group from the protein surface, and RA is the relative accessibility (RA) at the atomic level. RAD defines the composite information that combines the depth and exposure of a chemical group and can be calculated by using, for instance, the programs SADIC and POPS (Puglisi et al., Reference Puglisi, Brylski, Alfano, Martin, Pastore and Temussi2020). The SADIC algorithm calculates the intersection between the molecular volume and spheres centred on the atoms whose depth is quantified. This parameter yields how deeply the chemical group lies within the protein and measures its distance from the protein surface. The lower the value of the Depth parameter (D), the more buried the residue: residues with a D-value lower than 0.50 are considered to be buried. The program PopS yields instead relative accessibility (RA) at the atomic level: Q(SASA) is the ratio between the exposed surface of a nitrogen atom with respect to the SASA of the entire residue. The authors found that, by averaging only the residues with RAD <0.1, they could find a much better agreement with the far-UV CD data, a technique that provides only bulk information. In this way, Puglisi et al. (Reference Puglisi, Brylski, Alfano, Martin, Pastore and Temussi2020) demonstrated that the choice of the reporter is crucial for the reliable measurement of protein stability and also that proper signal averaging is essential. This study opened new directions to explore protein stability at the residue level.
Comparing unfolded states
The possibility of studying unfolding caused by different agents on the same protein called for a characterization of the cold and heat unfolded states. Adrover et al. (Reference Adrover, Esposito, Martorell, Pastore and Temussi2010, Reference Adrover, Martorell, Martin, Urosev, Konarev, Svergun, Daura, Temussi and Pastore2012) achieved virtually full assignment of Yfh1 at both −1 and 50 °C, showing that at these temperatures, below the cold and above the heat denaturation transitions, the protein has all the features of an unfolded protein. The authors found that cold unfolded Yfh1 retains some local, residual secondary structure but this does not reflect the secondary structural features of the folded species. The same laboratory (Adrover et al., Reference Adrover, Martorell, Martin, Urosev, Konarev, Svergun, Daura, Temussi and Pastore2012) deepened the comparison of the unfolded states at high and low temperatures by integrating small-angle X-ray Scattering (SAXS), NMR, and molecular dynamics data. SAXS measurements indicated that, despite the apparent local structural similarity observed by NMR, the two cold and heat equilibrium states of Yfh1 have a subtle but clear difference in size and compactness distribution. A detailed comparison of the chemical shifts and temperature coefficients revealed that the amide protons experience a different degree of solvation at high and low temperatures which indicated a higher degree of hydration at low temperatures (Adrover et al., Reference Adrover, Martorell, Martin, Urosev, Konarev, Svergun, Daura, Temussi and Pastore2012). This is in agreement with the model of cold denaturation formulated by Privalov (Reference Privalov1990).
Aznauryan et al. (Reference Aznauryan, Nettels, Holla, Hofmann and Schuler2013) also used Yfh1 to compare low- and high-temperature unfolded states directly on the same protein. Using single-molecule Förster resonance energy transfer (FRET) experiments, they found continuous compaction of the unfolded protein from 1 to 47 °C, with a minor re-expansion at higher temperatures. Possible discrepancies between FRET and SAXS data would be only apparent, and mainly based on the wrong idea that it is possible to correlate directly the quantities Re (the distance between donor and acceptor) and Rg (the gyration radius) obtained from FRET and SAXS respectively (Riback et al., Reference Riback, Bowman, Zmyslowski, Knoverek, Jumper, Hinshaw, Kaye, Freed, Clark and Sosnick2017). As illustrated by Fuertes et al. (Reference Fuertes, Banterle, Ruff, Chowdhury, Pappu, Svergun and Lemke2018): ‘FRET data cannot be used to extract Rg without making a series of simplifying assumptions about the connections between Rg and Re and the converse is true of SAXS data. Indeed, much of the conflict in inferences drawn from SAXS versus FRET originates from the fact that the two methods provide access to different quantities’. To describe the unfolded states at low and high temperatures in a simplistic way, we might say that the cold unfolded state is characterized by a large Re corresponding to a minimum possible volume. As the temperature is increased, Re becomes smaller but the corresponding volume increases as a consequence of progressive (disordered) folding.
Taken together, these results provided for the first time a detailed characterization and comparison of the heat and cold denaturation states highlighting their differences.
Yet another unfolded state: pressure unfolding
More recently, Yfh1 has been used to study the effects of pressure (Pastore and Temussi, Reference Pastore and Temussi2023; Roche et al., Reference Roche, Royer and Roumestand2017). This denaturing agent is interesting for at least three independent but related reasons: (i) pressure variations allow the exploration of the whole phase diagram of protein unfolding; (ii) under high pressure, stable proteins unfold without the need for chaotropic agents (e.g. urea or guanidinium) which may act as co-solvents and perturb the chemical environment and the solvation state of the protein (Pastore and Temussi, Reference Pastore and Temussi2023); (iii) high-pressure increases the temperature of cold denaturation and at the same time reduces the freezing point of water (Smeller, Reference Smeller2002), thus making cold denaturation studies more approachable even for very stable proteins.
High-pressure studies of Yfh1 were carried out both by fluorescence and NMR measurements, which provided complementary information (Puglisi et al., Reference Puglisi, Cioni, Gabellieri, Presciuttini, Pastore and Temussi2022). Fluorescence spectroscopy using the intrinsic fluorescence of the two tryptophan residues of Yfh1 allowed the determination of the phase diagram of unfolding as a function of pressure (1–5000 bar) and temperature 278–313 K (5–40 °C), both in the absence and in the presence of fold stabilizers (Puglisi et al., Reference Puglisi, Cioni, Gabellieri, Presciuttini, Pastore and Temussi2022). As expected, Yfh1 showed a remarkable sensitivity to pressure with a midpoint of unfolding already occurring at pressures around 100 bar at room temperature. These pressures should be compared with the midpoint pressure for other model globular proteins, such as the immunoglobulin-like module of titin I27 or a hyperstable variant of Staphylococcus nuclease, which require pressures as high as 2–3 kbar to unfold and the presence of small concentrations of guanidinium chloride (Herrada et al. Reference Herrada, Barthe, Vanheusden, DeGuillen, Mammri, Delbecq, Rico and Roumestand2018; Roche et al., Reference Roche, Caro, Norberto, Barthe, Roumestand, Schlessman, Garcia and Royer2012).
More recently, NMR measurements were used to get residue-specific information on the unfolding events (Roumestand et al., Reference Roumestand, Dudas, Puglisi, Calio, Barthe, Temussi and Pastoresubmitted). On the basis of the average chemical shifts of the resonances in the HSQC spectra of the three unfolded states, it was demonstrated that the Yfh1 pressure unfolded state shares features closer to those of the low than of the high temperature-unfolded state, suggesting a somewhat similar mechanism. Both the spectra of the pressure and cold-denatured species have in fact an overall low-field chemical shift as compared to the high-temperature unfolded spectrum, indicating de-shielding of the amide protons. A similar effect of pressure on the chemical shifts and a closer similarity between cold and pressure denaturation has also been reported for the cold shock protein B from Bacillus subtilis (BsCspB) (Berner and Kovermann, Reference Berner and Kovermann2024) and ubiquitin (Vajpai et al., Reference Vajpai, Nisius, Wiktor and Grzesiek2013). This conclusion agrees with Privalov’s theory (1979, 1982, 1990) according to which cold denaturation would depend on a higher affinity of water for apolar groups and on hydrogen bonding with the solvent, while heat denaturation would result from increasing molecular motions.
Analysis of the pressure-induced unfolding at different temperatures also allowed the determination of the hierarchical mechanism of Yfh1 unfolding using an approach introduced by C. Royer and cow (Roche et al., Reference Roche, Caro, Norberto, Barthe, Roumestand, Schlessman, Garcia and Royer2012). In this method, the probability of observing contacts between residues in the folded state is roughly given by the geometric mean of the fractional probabilities of the two residues at a given condition (Fossat et al., Reference Fossat, Dao, Jenkins, Dellarole, Yang, McCallum, Garcia, Barrick, Roumestand and Royer2016). The contacts above a certain threshold are compared with the contact map calculated from the coordinates of the folded protein. This is a rough but cunning way to get a qualitative idea of the regions of a protein that unfold first and if the process is cooperative. The authors observed that, near the temperature of maximal stability, pressure unfolding is highly cooperative, close to a global two-state equilibrium between the folded and unfolded populations of the protein (Figure 7). At temperatures above and below the temperature of maximal stability, unfolding becomes less cooperative at low pressures, whereas, at higher pressures, it is again highly cooperative at all temperatures. At moderate pressures and low temperatures, partial unfolding concerns primarily the N-terminal helix and the first strands of the ß-sheet, where the negatively charged cluster is (Bitonti et al., Reference Bitonti, Puglisi, Meli, Martin, Colombo, Temussi and Pastore2022; Sanfelice et al., Reference Sanfelice, Morandi, Pastore, Niccolai and Temussi2015). At high temperatures, unfolding starts at the C-terminal helix. This behaviour suggested a synergic mechanism between pressure- and temperature-induced denaturation in which pressure destabilization helps to unveil the hierarchical events of cold- and heat-induced unfolding. We could say that, metaphorically speaking, it is as if there were two ‘doors’ on the structure of the protein that promotes unfolding, one for the cold temperature, the other for the hot one. This is in perfect agreement with our previous studies that had shown that unfolding at low temperatures is mediated by the negatively charged frustration caused by residues in the N-terminus of the protein, whereas heat temperature unfolding is strongly dependent on the C-terminal mediated opening of the hydrophobic core.

Figure 7. Summary of the results obtained by recording thermal unfolding by NMR at progressively increasing pressures. At each data point, the probabilities of contacts calculated as a function of the peak intensities are calculated and compared to the contact map from the folded structure. Loss of all contacts is highly cooperative at high pressure. At low pressures (50–150 bar), the process is less cooperative, especially at low temperatures. It is clear that loss of contacts involves first the residues in the N-terminal helix and the beginning of the sheet at low temperature, whereas the first contacts that disappear at high temperature are those around the C-terminus. This behaviour is symbolised in the figure by a door sandwiched by the structures of Yfh1 in two orthogonal views to show the regions involved.
Overall, these studies at high pressure provided new and important information on the mechanisms of pressure unfolding of proteins and the relation between cold denaturation and other types of unfolding.
The kinetics of folding of Yfh1
Bonetti et al. (Reference Bonetti, Toto, Giri, Morrone, Sanfelice, Pastore, Temussi, Gianni and Brunori2014) used Yfh1 to consider a different aspect linked to protein folding and extensively characterized the temperature and urea dependence of the kinetics of folding of yeast frataxin both in the absence and in the presence of stabilizing salts. Their temperature-jump experiments between 288 and 333 K supported a simple two-state process involving a single energy barrier. In the presence of urea as a chaotropic denaturant in the pH range 4–9, the authors observed instead that the main rate-limiting step conformed to a smooth broad-free energy barrier. This meant that, while the data recorded in the absence of denaturant could be approximated by a simple two-state model for the temperature dependence of the folding and unfolding rate constants, the data in urea are less cooperative.
The observation of a smooth broad free energy barrier is interesting because it provided valuable information about the transition state: it indicated that Yfh1 belongs to a subset of globular proteins, which includes U1A and azurin, that have a broad energetic barrier to folding, implying a malleable structure. This is at variance with many globular proteins that appear to fold via a robust transition state, whose structure is not affected by changes in experimental conditions or denaturant concentration. This behaviour has been ascribed by Wolynes and co-workers to the presence of energetic frustration in the native state ensemble in agreement with the results by Sanfelice et al., Reference Sanfelice, Morandi, Pastore, Niccolai and Temussi2015. Bonetti et al. also suggested that the possibility of studying the residual secondary structure of the cold- and heat-denatured states (Adrover et al., Reference Adrover, Esposito, Martorell, Pastore and Temussi2010, Reference Adrover, Martorell, Martin, Urosev, Konarev, Svergun, Daura, Temussi and Pastore2012) provides a unique tool to study Ptitsyn’s theory in which an overall three-dimensional collapse precedes the tight packing of the side chains implying the existence of intermediates with residual secondary structures (Ptitsyn, Reference Ptitsyn1995).
Further analysis of the transition-state structure revealed that the regions involved in the folding process are highly aggregation-prone, while functionally important regions are partially misfolded in the transition state but highly resistant to aggregation (Gianni et al., Reference Gianni, Camilloni, Giri, Toto, Bonetti, Morrone, Sormanni, Brunori and Vendruscolo2014). These results indicated that the competition between folding and function is at the very basis of the marginal stability of this protein and provided the grounds for a more recent study in which the folding of Yfh1 was compared to that of the human orthologue (Pietrangeli et al., Reference Pietrangeli, Marcocci, Pennacchietti, Diop, Di Felice, Pagano, Malagrinò, Toto, Brunori and Gianni2024). The comparison suggested a weak native bias at the early stages of folding, with observed pathways converging only when the native state is approached. The authors concluded that consolidation of the folding nucleus of this protein is slightly decoupled from a general collapse of the polypeptide chain, which appears to precede nucleation according to Ptitsyn’s theory (1995).
Stability or resilience? chance or necessity?
Yfh1 is a protein with very unique properties as it can denature under several different conditions without ever degrading into an irreversible aggregate state, provided suitable amounts of reducing agents are present. In the original article discussing the properties of this protein, Pastore, Martin, et al. (Reference Pastore, Martin, Politou, Kondapalli, Stemmler and Temussi2007) went above the temperature of heat unfolding and below that of cold denaturation several times using the same protein sample but, after these operations, the NMR spectrum at room temperature remained indistinguishable from the original spectrum. Again, Martin et al. (Reference Martin, Esposito, De Los Rios, Pastore and Temussi2008) went several times across both unfolding temperatures in solutions containing methanol, ethanol, and trifluoroethanol at concentrations comprised between 0 and 15%. Also in these cases, going back to room temperature yielded the same NMR spectrum. The unfolding transitions of Yfh1 were also mostly reversible at high pressure: after recording spectra at different temperatures, the spectrum collected after returning to room temperature and pressure lost only a minor fraction of its intensity (Puglisi et al., Reference Puglisi, Brylski, Alfano, Martin, Pastore and Temussi2020; Roumestand et al., Reference Roumestand, Dudas, Puglisi, Calio, Barthe, Temussi and Pastoresubmitted). Also interesting is that, at pH values around neutrality, Yfh1 can be concentrated at least to the low millimolar range without aggregation. We can thus conclude that Yfh1, although only a marginally stable protein, is also characterized by a high resilience, meaning with this term the process of adapting well to significant sources of stress. From this point of view, resilience might be considered as a specific aspect of stability. For a protein in vivo, it is probably more important to have a high resilience than a large (static) stability, which by itself might prevent resilience.
Indeed, Yfh1 is the first natural protein found to show cold denaturation above the freezing point of water under quasi-physiological conditions, where ‘quasi’ refers to the fact that cold denaturation is observed only at low ionic strength. It is worth wondering at this point how such a marginally unstable protein can survive in nature and whether these properties are needed for its physiological functions. Low-salt conditions might be difficult to find in a natural environment, especially for intracellular proteins mainly localized in the mitochondrion. We could thus easily argue that Yfh1 will never experience in nature the conditions we have in the test tube. However, two aspects need to be considered: first, all the conditions chosen for in vitro unfolding studies are rather unrealistic. Hen egg-white lysozyme or human ubiquitin will for instance never experience 1000 bar pressures. Yet, extreme conditions help us to observe processes usually less evident. Second, the observation of what happens to Yfh1 at low salt levels immediately suggests that, unless intrinsically disordered which is what the protein is not, there must be factors that stabilise the protein in nature. We now know that the protein binds to iron and this is a factor of strong stabilization. It also binds to other protein partners.
Conclusions: Yfh1 and beyond
In conclusion, in the previous paragraphs, we have discussed several examples in which Yfh1 has been used as a ‘tool’ to study the influence of several environmental conditions on protein stability. Yfh1 has proven to be a ductile and versatile system that can assist in reconsidering standing unanswered questions with a new fresh perspective. From this model system, we have learned important lessons, some of which have broken established paradigms, on the very nature of protein stability and the factors influencing it. As a result, it would be fair to conclude that stability is not just a number, but rather an ensemble of conditions characterizing a protein. Likewise, the protein unfolding state is far from being unique and has instead specific features characteristic of the environmental conditions. Future questions that Yfh1 can still help us to address include understanding the kinetics of protein unfolding under different conditions and rules that determine protein aggregation rather than unfolding. We expect that Yfh1 can provide ‘the’ perfect system for the study of protein unfolding which should become more generally exploited. Another urgent open question is also to demonstrate how much of what we have learned from Yfh1 is exportable to other proteins. While some progress along these lines has already been made by transforming the stable bacterial frataxin into a cold denaturing protein (Bitonti et al., Reference Bitonti, Puglisi, Meli, Martin, Colombo, Temussi and Pastore2022), we now need to validate our working hypotheses using completely unrelated proteins with different folds. It will also be important to compare the lessons learned on Yfh1 on the other few systems, most of which are not natural proteins, that are known to give a detectable cold denaturation. We believe that the knowledge gained will ultimately allow us to better understand the physical forces holding together proteins and the rules that prevent their unfolding and/or misfolding.
Acknowledgements
We would like to thank all the people in our group and the collaborators who have contributed to the study of Yfh1 through the years.
Financial support
The authors declare that no funds, grants, or other support were received during the preparation of this manuscript.
Competing interest
The authors declare to have none.






 Open access
Open access