


Pre- and postnatal malnutrition may have serious consequences for neuronal development and growthReference Guesry1, Reference Morgane, Austin-LaFrance, Bronzino, Tonkiss, az-Cintra, Cintra, Kemper and Galler2. The impact of vitamin C deficiency, beyond that of scurvy, has not been studied in detail. Vitamin C, for example, is known to play an important role in maintaining brain functionReference Rice3. The brain is particularly susceptible to oxidative damage and therefore highly dependent on proper maintenance of redox homeostasis, especially during development, when brain metabolism and growth are maximalReference Rougemont, Do and Castagne4. An imbalance in redox homoestasis has also been implicated in neurodegenerative diseases of ageing and other neurological disorders such as schizophreniaReference Bains and Shaw5–Reference Schulz, Lindenau, Seyfried and Dichgans8.
It has been known for a while that vitamin C is preferentially retained in the brain during deficiencyReference Kuo, Yonehara and Yoshida9. As established recently, this is due to the high-affinity sodium ascorbate cotransporter SVCT2Reference Sotiriou, Gispert and Cheng10. Experiments with homozygous SVCT2-knockout mice have shown that this transporter is essential for perinatal survivalReference Sotiriou, Gispert and Cheng10, indicating that vitamin C is crucial for early brain development, although the precise mechanism of death is unknown. Moreover, studies of combined vitamin C and E deficiency in guinea pigs have revealed extensive neuronal damage despite a relatively modest increase in oxidative stress in the brainReference Burk, Christensen, Maguire, Austin, Whetsell, May, Hill and Ebner11, Reference Hill, Montine, Motley, Li, May and Burk12.
Unfortunately, neonatal vitamin C deficiency is fairly common. In a study with 127 pregnant Brazilian women, it was found that 40 % of the smokers and 27 % of the non-smokers had hypovitaminosis C (i.e. a plasma concentration < 23 μmol/l) and that this condition was passed on to their fetusesReference Madruga de Oliveira, de Carvalho Rondo and Barros13. Major epidemiological studies have shown that this prevalence of hypovitaminosis C can largely be extended to the Western worldReference Lykkesfeldt, Halliwell and Poulsen14. Vitamin C deficiency due to inadequate perinatal feeding (e.g. with pasteurized milk) is also fairly commonReference Ratanachu-Ek, Sukswai, Jeerathanyasakun and Wongtapradit15.
Like man, guinea pigs completely depend on dietary vitamin CReference Chatterjee16, Reference Nandi, Mukhopadhyay, Ghosh, Chattopadhyay and Chatterjee17. In the present study, we used newly weaned guinea pigs to test if neonates are susceptible to vitamin C deficiency and if even small changes in antioxidant status in the brain result in deleterious events as measured by oxidative damage.
Methods
Animals and study design
The study was approved by the Danish Animal Experimentation Inspectorate. Dunkin Hartley guinea pigs were born in our animal facility by breeder guinea pigs obtained from Statens Serum Institut (Allerød, Denmark), that had been maintained at the animal facility for at least 2 weeks. After 2 d, the neonates were taken from their mothers and randomized into two weight-matched groups (control group, n 8; deficient group, n 12). All animals were housed in plastic cages (four per cage) and fed pathogen-free standard diets (Altromin International, Lage, Germany). The diets were essentially identical except for vitamin C content. Control animals were placed on a normal guinea pig diet (Diet #3010, 1036 mg vitamin C/kg)18, while deficient animals were supplied with a diet low in vitamin C (Diet #2010, 36 mg vitamin C/kg)19. The dietary regimen was continued for about 3 weeks. Animals were checked daily by educated staff and weighed twice a week (Fig. 1). No clinical signs of scurvy were observed during the experiment.
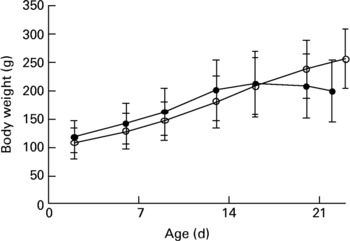
Fig. 1 Weights of 2-d-old neonatal guinea pigs maintained on either a control diet (○) or a vitamin C-deficient diet (●) for 3 weeks. No significant difference was observed over the course of the study period. Values are means with standard deviations depicted by vertical bars.
After 3 weeks, the animals were anaesthetized using pentobarbital (55 mg/kg containing 2 % lidocain, by intraperitoneal injection). Heparin (500 units; 100 μl) was injected intracardially using a 27G needle and after 1 min a 2 ml blood sample was obtained by cardiac puncture using a 18G needle while carefully avoiding haemolysis. The animals were perfused with 100 ml ice-cold PBS, after which liver and brain were removed and immediately frozen at − 80°C until further analysis. Blood samples were immediately centrifuged (2000g, 5 min, 4°C). One aliquot (200 μl) of plasma was acidified with an equal volume of 10 % meta-phosphoric acid containing 2 mm-EDTA, briefly vortex mixed and frozen at − 80°C for ascorbate (Asc) and dehydroascorbic acid analysis. The remaining plasma was stored neat in aliquots at − 80°C.
Biochemical analyses
Asc and dehydroascorbic acid in meta-phosphoric acid-stabilized plasma and tissues were analysed by HPLC with coulometric detection as described previouslyReference Lykkesfeldt20, Reference Lykkesfeldt, Maines, Costa, Hodson and Reed21. Because of the very low vitamin C levels in plasma of animals fed with the deficient diet, dehydroascorbic acid was below the detection limit of this assay. α-Tocopherol and γ-tocopherol were analysed by HPLC with amperometric detection as describedReference Sattler, Mohr and Stocker22. Glutathione was measured as described by Hissin & HilfReference Hissin and Hilf23. Superoxide dismutase activity was quantified by the pyrogallol methodReference Marklund and Marklund24. Plasma oxidizability was quantified essentially as described by Kontush & BeisiegelReference Kontush and Beisiegel25 except that lag time was objectively based on a sigmoidal curve-fitting model with subsequent calculation of the inflexion. The intercept between the baseline and inflexion was used as lag time.
Malondialdehyde (MDA) was used as an index of lipid oxidation and was assessed by thiobarbituric acid (TBA) derivatization, followed by specific quantification of the genuine MDA(TBA)2 adduct by HPLC with fluorescence detection as described previouslyReference Lykkesfeldt26. Protein carbonyls were measured using the ZenTech ELISA kit (Alexis Corporation) based on the method by Buss & WinterbournReference Buss and Winterbourn27. Oxidatively modified DNA as measured by 8-oxo-deoxyguanosine content was quantified by HPLC with electrochemical detection as describedReference Gedik and Collins28 and expressed per 106 unmodified guanosine residues. Base excision repair was estimated by the nicking technique as described earlierReference Riis, Risom, Loft and Poulsen29. Protein concentrations were measured by using the Bradford methodReference Bradford30.
Statistical analysis
Effects were analysed by one-way ANOVA followed by post hoc t-tests. A two-tailed P value less than 0·05 was considered statistically significant. All data are reported as means and standard deviations.
Results and discussion
In the present study, we examined the effect of vitamin C deficiency on liver and brain redox homeostasis in weanling guinea pigs. We were particularly interested in the brain, since it is highly susceptible to oxidative damage and known to accumulate high concentrations of Asc, the reduced and antioxidative form of vitamin C. Although no significant differences in growth were observed over the course of the study (Fig. 1), the deficient guinea pigs showed clear signs of growth arrest after 3 weeks and would have developed scurvy and died had the experiment been allowed to continue. Apart from its antiscorbutic effect, vitamin C plays a pivotal role in redox homeostasisReference Rice3, Reference Buettner, Schafer, Asard, May and Smirnoff31, Reference Rice and Russo-Menna32.
Weanling guinea pigs fed for 3 weeks on a vitamin C-deficient diet displayed severe depletion of plasma and liver Asc, showing less than 1 % Asc compared to control animals (P < 0·001; Table 1). In contrast, the depletion of Asc in the brain was only about 70 %. This preferential preservation of vitamin C in the brain is in agreement with other studiesReference Kuo, Yonehara and Yoshida9, Reference Hughes, Hurley and Jones33 and is most likely the result of active transport of vitamin C across the blood–brain barrier.
Table 1 Biomarkers of oxidative stress and damage in plasma, liver and brain of weanling guinea pigs after 3 weeks on a vitamin C-deficient diet compared to animals on a control diet
(Mean values and standard deviations)

dG, unmodified guanosine residues.
* Compared to control group.
† Control, n 4; deficient, n 8.
Asc depletion in plasma and liver was accompanied by a general change in the antioxidant status. Thus, vitamin C deficiency caused a decrease of α- and γ-tocopherol in plasma and liver, and resulted in lower glutathione levels and superoxide dismutase activity in the liver, suggesting a collapse of the redox homeostasis in this tissue (Table 1). In contrast, Asc was the only antioxidant in the brain that was significantly decreased. The significant 2·5-fold increase in dehydroascorbic acid observed in vitamin C-deficient animals (P = 0·034) is indicative of elevated oxidative stress in the brain. Thus, increased antioxidant action of Asc combined with inadequate recycling capacity for vitamin C results in increased turnover as recognized by the increased presence of dehydroascorbic acid.
Vitamin C deficiency was also associated with increased oxidative damage. In plasma, lipid oxidation was significantly increased in vitamin C-deficient animals (P = 0·013) and the resistance to lipid oxidation (lag time) was significantly diminished to less than one-quarter of that of animals fed with the normal diet (P < 0·001). In the liver, protein oxidation was increased (P = 0·011) while lipid and DNA oxidation were unchanged.
As with the antioxidants, the pattern of oxidative damage was different in the brain. Here, MDA was increased by 130 % in deficient animals compared to that of controls (P < 0·001). In contrast, protein oxidation was significantly lower (P = 0·003). Whether this is related to vitamin C's role in protein synthesis is not known. With regard to DNA damage, both oxidatively damaged DNA and DNA repair capacity was assessed, the latter as 8-oxo-deoxyguanosine glycosylase activity. Since de novo synthesis of DNA is only possible during cell proliferation, the capacity to repair damaged DNA is of major importance for the ability of the cells to survive an oxidative insult. In the brain, DNA incision repair was 38 % higher in deficient animals compared to controls (P = 0·014), suggesting an up-regulation of DNA repair in response to the increased oxidative stress. This response was apparently adequate, since 8-oxo-deoxyguanosine did not significantly increase in the vitamin C-deficient animals (P = 0·13).
Recently, it was reported that in vitro-cultured cells do not respond to oxidative DNA damage by inducing repairReference Bercht, Flohr-Beckhaus, Osterod, Runger, Radicella and Epe34. The authors concluded that DNA damage per se does not induce DNA repairReference Bercht, Flohr-Beckhaus, Osterod, Runger, Radicella and Epe34. However, it is possible that DNA repair was already maximally induced in their in vitro system, or that induction was no longer possible due to cell transformation. Our in vivo data, however, suggest that either the reduced levels of antioxidants per se or a secondary effect of such depletion indeed do induce DNA repair in vivo.
Neonatal guinea pigs appear to be more susceptible to malnutrition compared to their older counterparts, possibly due to their limited reserve of micronutrients and their rapid growth. This became apparent in the present study as the neonates began suffering growth arrest and even weight loss, usually among the first signs of emerging lameness in the hind limbs and clinical scurvy, already after 3 weeks (Fig. 1), while in older animals (e.g. 3 months old) the same condition is normally reached only after more than 5 weeks on a deficient dietReference Lykkesfeldt35. Thus, neonates appear to be particularly susceptible to vitamin C deficiency.
Conclusion
The present results show that in weanling guinea pigs, vitamin C deficiency results in altered brain redox homeostasis and increased lipid oxidation. Preferential preservation of vitamin C in the brain over other tissues, antioxidant function of Asc and induction of DNA incision repair provide some protection against oxidative damage to DNA and proteins. To strengthen the link between oxidative stress and disease, future studies should include the evaluation of brain injury. Also, the expression of SVCT2 in the brain during vitamin C deficiency may be of importance in understanding the preferential preservation of vitamin C in the brain.
Acknowledgements
Annie B. Kristensen, Jytte Nielsen, Corinne Siegenthaler, Jane Povlsen and the KVL animal facility are thanked for excellent technical assistance. This study was supported by National Research Council grant 21-01-0525 to J. L. The laboratory of S. C. is supported by grants from the Swiss National Science Foundation (3100-108236) and National Institute of Neurological Disorders and Stroke (R01 NS33997).


