



Introduction
Parahibbingite corresponds to the β-form of ferrous-hydroxychloride, which was first synthesised and described by Oswald and Feitknecht (Reference Oswald and Feitknecht1964); since then, it has often been reported as a Cl-rich corrosion product of archaeological iron artefacts, and it has been the subject of numerous studies aimed at the conservation of archaeological objects (e.g. Réguer et al., Reference Réguer, Dillmann, Mirambet and Bellot-Gurlet2005, Reference Réguer, Dillmann and Mirambet2007a; Azoulay et al., Reference Azoulay, Conforto, Refait and Rémazeilles2013).
Natural and anthropogenic specimens of the β-form of ferrous hydroxychloride typically occur as microcrystalline masses included in thin corrosion layers at the metal/oxide interface associated with akaganeite (β-FeOOH) (Réguer et al., Reference Réguer, Mirambet, Rémazeilles, Vantelona, Kergourlaya, Neff and Dillmann2015), whereas synthetic samples are polycrystalline powders (Fujihala et al., Reference Fujihala, Hagihala, Zheng and Kawae2010).
The recent finding of microcrystalline aggregates among the alteration products recovered from a drill core from the Karee platinum mine in the Bushveld Complex, South Africa, allowed Koděra et al. (Reference Koděra, Majzlan, Pollok, Kiefer, Šimko, Scholtzová, Luptáková and Cawthorn2022) to get parahibbingite approved by the International Mineralogical Association as a new species. The new mineral occurred as fine-grained rims at the contact between orthopyroxene and talc, as a result of late hydrothermal alteration of pyroxenite.
In addition to the β-phase, ferrous hydroxychloride exhibits two other polymorphs: α and γ (Oswald and Feitknecht, Reference Oswald and Feitknecht1964). The metastable α-Fe2(OH)3Cl (space group P $\bar{3}$ m1) has never been found in Nature (Koděra et al., Reference Koděra, Majzlan, Pollok, Kiefer, Šimko, Scholtzová, Luptáková and Cawthorn2022). The natural γ-phase (space group Pnam), named hibbingite, was first described by Saini-Eidukat et al. (Reference Saini-Eidukat, Kucha and Keppler1994) in a partially serpentinised troctolite from the Duluth Complex, Minnesota, USA, and since then it has been occasionally reported in both ore deposits and in the alteration crust of meteorites (e.g. Saini-Eidukat et al., Reference Saini-Eidukat, Rudashevsky and Polozov1998; Pekov et al., Reference Pekov, Perchiazzi, Merlino, Kalachev, Melrini and Zadov2007; Zubkova et al., Reference Zubkova, Pekov, Sereda, Yapaskurt and Pushcharovsky2019). However, due to its microcrystalline nature, it can be difficult to distinguish between β- and γ-Fe2(OH)3Cl. Furthermore, the name hibbingite has often been used incorrectly to refer to the β-phase (e.g. Buchwald and Koch, Reference Buchwald and Koch1995; Simon et al., Reference Simon, Cibin, Robbins, Day, Tang, Freestone and Schofield2018) and it is likely that some samples identified as hibbingite may actually be parahibbingite (Koděra et al., Reference Koděra, Majzlan, Pollok, Kiefer, Šimko, Scholtzová, Luptáková and Cawthorn2022).
m1) has never been found in Nature (Koděra et al., Reference Koděra, Majzlan, Pollok, Kiefer, Šimko, Scholtzová, Luptáková and Cawthorn2022). The natural γ-phase (space group Pnam), named hibbingite, was first described by Saini-Eidukat et al. (Reference Saini-Eidukat, Kucha and Keppler1994) in a partially serpentinised troctolite from the Duluth Complex, Minnesota, USA, and since then it has been occasionally reported in both ore deposits and in the alteration crust of meteorites (e.g. Saini-Eidukat et al., Reference Saini-Eidukat, Rudashevsky and Polozov1998; Pekov et al., Reference Pekov, Perchiazzi, Merlino, Kalachev, Melrini and Zadov2007; Zubkova et al., Reference Zubkova, Pekov, Sereda, Yapaskurt and Pushcharovsky2019). However, due to its microcrystalline nature, it can be difficult to distinguish between β- and γ-Fe2(OH)3Cl. Furthermore, the name hibbingite has often been used incorrectly to refer to the β-phase (e.g. Buchwald and Koch, Reference Buchwald and Koch1995; Simon et al., Reference Simon, Cibin, Robbins, Day, Tang, Freestone and Schofield2018) and it is likely that some samples identified as hibbingite may actually be parahibbingite (Koděra et al., Reference Koděra, Majzlan, Pollok, Kiefer, Šimko, Scholtzová, Luptáková and Cawthorn2022).
Nevertheless, Réguer et al. (Reference Réguer, Mirambet, Rémazeilles, Vantelona, Kergourlaya, Neff and Dillmann2015) were able to recognise coexisting β and γ phases occurring in archaeological iron corroded in a marine environment by means of powder X-ray diffraction. In addition, these two phases can be easily distinguished via Raman spectroscopy (Koděra et al., Reference Koděra, Majzlan, Pollok, Kiefer, Šimko, Scholtzová, Luptáková and Cawthorn2022).
The recent occurrence of exceptionally well-crystallised parahibbingite associated with a fragment of the Muonionalusta iron meteorite from northernmost Sweden (Wickman, Reference Wickman1964; Buchwald, Reference Buchwald1975) allowed us to carry out a single-crystal X-ray diffraction study and collect high-quality micro-Raman data from the mineral.
Occurrence
The Muonionalusta meteorite consists of a collection of a large number of fragments, ranging from 0.04 up to 1200 kg, found in a wide area (25 × 15 km) centred around lake Kitkiöjärvi, Pajala District, Norrbotten County, Sweden. It is deemed that the original body of the Muonionalusta meteorite did not hit the ground as a single impactor, but exploded mid-air, creating a meteorite shower (Wickman, Reference Wickman1964; Lagerbäck and Wickman, Reference Lagerbäck and Wickman1997). The meteorite fragments have experienced at least one glacial period (the Weichselian, 115 to 12 ka), and associated glacial transport processes could be responsible, along with the impact dynamic, for the considerable size of the present distribution area (Hättestrand, Reference Hättestrand2009).
The Muonionalusta meteorite belongs to group IVA and consists mainly of Fe–Ni alloys forming a fine octahedrite texture and containing polycrystalline troilite nodules and minor amounts of daubréelite, chromite and schreibersite (Buchwald, Reference Buchwald1975). It is the first iron meteorite in which stishovite has been reported, probably formed during shock metamorphism in space of primary tridymite (Holtstam et al., Reference Holtstam, Broman, Söderhielm and Zetterqvist2003).
Fragments of the meteorite are generally found at different depths in tillite beds, sometimes intermingled with glaciofluvial sediments, and are usually covered by a crust of rusty material, which commonly includes fragments of the surrounding soil. The crust is populated by a wide variety of alteration products, mainly iron oxides, oxyhydroxides and hydroxychloride, such as magnetite, maghemite, goethite, akaganeite, lepidocrocite, and the recently approved mineral muonionalustaite [Ni3(OH)4Cl2⋅4H2O] (Holtstam et al., Reference Holtstam, Bindi, Karlsson, Söderhielm and Zetterqvist2020).
Parahibbingite has been found in a small cavity of rust crust on a 9 × 7 × 4 cm granitic rock fragment associated with the alteration crust of the meteorite (sample GEO-NRM #20060957). The fragment was in direct contact with a 22 kg specimen of the Muonionalusta meteorite, recovered from a depth of 1.4 m, ca 5 km NE of Kitkiöjärvi. The cavity was found to be filled with an unknown brownish liquid when it was opened in the field.
Parahibbingite forms well-developed crystals (transparent with a greenish tinge) up to 2 mm in size, and is covered by a reddish-brown rim of akageneite (Fig. 1). In the same cavity, brownish-orange hexagonal-shaped lamellae of an unknown Fe- and sulfate-bearing phase have been found. Despite their distinct morphology, these lamellae turned out to be a poorly crystallised phase which is still under investigation. It is likely that at least partly, some of the akaganeite in the present sample formed during atmospheric oxidising post-excavation conditions (since 2006).

Fig. 1. Mineral association consisting of parahibbingite crystals covered with a reddish-brown coating of akaganeite, and brownish-orange lamellae of an unknown, poorly crystallised phase. Field of view = 6 mm. Picture courtesy of Torbjörn Lorin, private collection.
X-ray crystallography and structure refinement
Several crystal fragments of parahibbingite, ranging from 0.1 up to 0.2 mm in size, were examined by means of a D8 Advance diffractometer equipped with a Photon III detector, using graphite-monochromatised MoKα radiation (λ = 0.71073 Å). The unit-cell values were nearly identical for all the crystals tested. Intensity data collection was carried out on the crystal showing the best diffraction quality [unit-cell parameters: a = 6.9362(4), c = 14.673(1) Å and V = 611.35(7) Å3]. Intensities were corrected for Lorentz and polarisation factors and absorption using the software package APEX3 (Bruker, 2016).
The merging factor in the Laue group $\bar{3}$ m (R int = 0.040), reflection conditions (hkl: –h+k+l = 3n) and the statistical tests on the distribution of |E| values (|E 2 – 1| = 0.917) suggested the space group R $\bar{3}$
m (R int = 0.040), reflection conditions (hkl: –h+k+l = 3n) and the statistical tests on the distribution of |E| values (|E 2 – 1| = 0.917) suggested the space group R $\bar{3}$ m, in accord with that determined for the synthetic samples. Therefore, the structure was refined starting from the atomic coordinates reported for synthetic β-Fe2(OH)3Cl (Réguer et al., Reference Réguer, Mirambet, Rémazeilles, Vantelona, Kergourlaya, Neff and Dillmann2015) using the program Shelxl-2013 (Sheldrick, Reference Sheldrick2015). The site occupancy factor (s.o.f.) at the cation sites was allowed to vary (Fe vs. structural vacancy for the two octahedral sites) using scattering curves for neutral atoms taken from the International Tables for Crystallography (Wilson, Reference Wilson1992). Two sites (Wyckoff positions 3b and 9e, labelled Fe1 and Fe2 respectively) were found to be fully occupied by Fe atoms and thus their occupancy was fixed in the successive least-squares cycles and an anisotropic model of the whole structure was refined.
m, in accord with that determined for the synthetic samples. Therefore, the structure was refined starting from the atomic coordinates reported for synthetic β-Fe2(OH)3Cl (Réguer et al., Reference Réguer, Mirambet, Rémazeilles, Vantelona, Kergourlaya, Neff and Dillmann2015) using the program Shelxl-2013 (Sheldrick, Reference Sheldrick2015). The site occupancy factor (s.o.f.) at the cation sites was allowed to vary (Fe vs. structural vacancy for the two octahedral sites) using scattering curves for neutral atoms taken from the International Tables for Crystallography (Wilson, Reference Wilson1992). Two sites (Wyckoff positions 3b and 9e, labelled Fe1 and Fe2 respectively) were found to be fully occupied by Fe atoms and thus their occupancy was fixed in the successive least-squares cycles and an anisotropic model of the whole structure was refined.
Convergence was achieved quickly to R 1 = 0.0335 for 320 observed reflections [F o2 > 4σ(F o2)] and 0.0501 for all 402 independent reflections. At this stage, the inspection of the ΔF-map showed a significant residual peak of ≃ 2 e –/Å3 at ~0.95 Å from the oxygen atom, that was attributed to an H atom. The introduction of this additional peak slightly improved the R indices [R 1 [F o2 > 4σ(F o2)] = 0.0331, R 1 = 0.0496]. Nevertheless, the U iso of the hydroxyl H atom had a too high value, so the H atom was refined using a riding model with U iso(H) = 1.5×U eq(O) and the O–H distance restrained to 0.90(2) Å.
Experimental details are reported in Table 1. Fractional atomic coordinates and anisotropic-displacement parameters are given in Table 2 and 3, respectively. Selected bond distances, angles and bond valence sums are given in Table 4. Further details of the crystal structure investigations and the list of the observed and calculated structure factors may be obtained from the crystallographic information file deposited with CCDC/FIZ Karlsruhe online deposition service [https://www.ccdc.cam.ac.uk/structures/ under the deposition number CSD-2192582] or with Supplementary material (see below).
Table 1. Data and experimental details.

R int = Σ|F o2 – F o2(mean)|/Σ [F o2]. GooF = S = {Σ[w(F o2 – F c2)2]/(n – p)}½, where n = no. of reflections, p = no. of refined parameters. R 1 = Σ||F o| – |F c||/Σ|F o|. wR 2 = {S[w(F o2 – F c2)2]/S[w(F o2)2]}½; w = 1/[σ2(F o2) + (0.340P)2] where P = [F o2 + 2 (F c2)]/3.
Table 2. Atoms, Wyckoff positions, atomic coordinates and atomic displacement parameters for the structure of parahibbingite.

* The isotropic displacement parameter of the H atom was fixed during the refinement (see text).
Table 3. Anisotropic displacement parameters for the structure of parahibbingite.

Table 4. Selected bond distances (Å), angles (°) and bond valence sums (BVS) for the structure of parahibbingite.

The mean quadratic elongation (λ) and the angle variance (σ2) were computed according to Robinson et al. (Reference Robinson, Gibbs and Ribbe1971); the bond valence sums (BVS) were calculated according to the parameters of Brese and O'Keeffe (Reference Brese and O'Keeffe1991).
Micro-Raman investigation
A micro-Raman spectrum of parahibbingite (Fig. 2) was collected from a polished crystal on a LabRAM HR 800 micro-spectrometer, using a 514 nm Ar-ion laser source at 1 mW power, a peltier-cooled (–70°C) CCD detector (Synapse), Olympus objective (magnification 100/ numerical aperture 0.9) and laser spot of ~3 μm. Spectral positions were corrected against the Raman band at 789 cm–1 of pure SiC-6H on {0001}. The spectral resolution is ~1 cm–1. Instrument control and data acquisition (range 100–4000 cm–1) were made with the LabSpec 5 software. The specimen showed no sign of degradation under the laser beam during measurement.

Fig. 2. Micro-Raman spectrum of parahibbingite obtained with a 514 nm laser.
In the spectrum obtained, Raman bands are detected at 3565, 3553, 810, 618, 428, 321, 202, 163 and 127 cm–1. The positions agree within ±5 cm–1 with data for β-Fe2(OH)3Cl obtained from the corrosion product of a 15th Century nail (Réguer et al., Reference Réguer, Neff, Bellot-Gurlet and Dillmann2007b) and with the recent data for parahibbingite (Koděra et al., Reference Koděra, Majzlan, Pollok, Kiefer, Šimko, Scholtzová, Luptáková and Cawthorn2022). Compared to previous measurements, our spectrum is free from contributions from admixed impurities and has a considerably lower noise level. Following the assignments of Réguer et al. (Reference Réguer, Neff, Bellot-Gurlet and Dillmann2007b), the two strongest bands at ~3660 cm–1 are related to OH-stretching vibration modes. Multiple bands in the region 300–700 cm–1 are ascribed to Fe–Cl and Fe–O stretching modes. The band at ~810 cm–1 belongs to a hydroxyl deformation mode (Réguer et al., Reference Réguer, Neff, Bellot-Gurlet and Dillmann2007b), whereas the one at 163 cm–1 corresponds to bending of O–Fe–O bonds. The relatively strong peak at 127 cm–1 is tentatively assigned to lattice modes.
As regards the two bands in the OH region, they also appear in the spectra of previous papers at almost the same positions. Réguer et al. (Reference Réguer, Neff, Bellot-Gurlet and Dillmann2007b) supposed that the two major hydroxyl stretching bands could be related to two crystallographically independent hydroxyl groups. For this reason, we carried out structural refinements at lower symmetries than R $\bar{3}$ m (starting from R $\bar{3}$
m (starting from R $\bar{3}$ ) and also tried to refine O-versus-Cl occupancies at both anionic positions, but no indication of possible mixed occupancies was found thus leading to the conclusion that there are not different hydrogen environments. Alternatively the split of vibration bands observed in the Raman spectrum could be related to chemical substitutions at the octahedral sites which would create slightly different surroundings, i.e. next-nearest-neighbour effects. However, from previous microchemical (SEM-EDS) data, we know that the maximum metal impurity is ca. 0.05 apfu of Co, and this is not expected to create a significant effect.
) and also tried to refine O-versus-Cl occupancies at both anionic positions, but no indication of possible mixed occupancies was found thus leading to the conclusion that there are not different hydrogen environments. Alternatively the split of vibration bands observed in the Raman spectrum could be related to chemical substitutions at the octahedral sites which would create slightly different surroundings, i.e. next-nearest-neighbour effects. However, from previous microchemical (SEM-EDS) data, we know that the maximum metal impurity is ca. 0.05 apfu of Co, and this is not expected to create a significant effect.
Description of the structure and discussion
The crystal structure of parahibbingite (Fig. 3) can be described as a stacking along the c axis of two different types of layers, made of octahedrally coordinated Fe2+ atoms. In one layer, iron cations are arranged in Kagomé planes [Fe2 atoms, Fig. 3a], while in the other layer, they are disposed in triangular planes [Fe1 atoms, Fig. 3b].
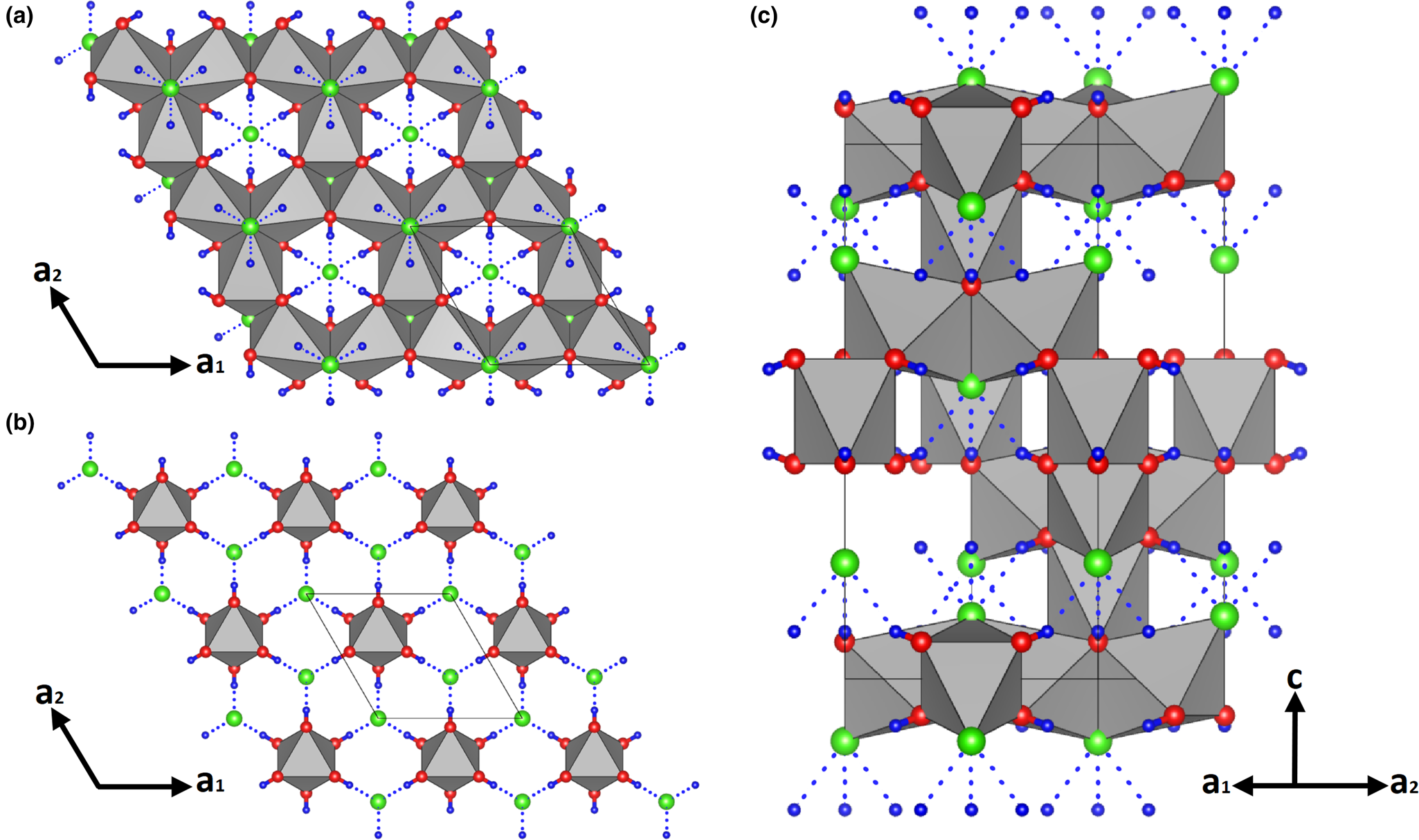
Fig. 3. The crystal structure of parahibbingite. (a) Kagomé planes and (b) triangular planes seen down the c axis. (c) View along the c axis. Colour scheme: Fe = grey; Cl = green; O = red; H = blue. H…Cl bridges are indicated as dashed lines. Drawn using Vesta (Momma and Izumi, Reference Momma and Izumi2011).
Fe1O6 octahedra are disjointed from each other, but they share O–O edges with Fe2O4Cl2 polyhedra of the upper and lower layers, while Fe2O4Cl2 octahedra are joined in interconnected triplets sharing a Cl–O edge. Each chlorine atom acts as acceptor of three H bonds from the oxygen atom (dotted lines in Fig. 3c), thus reaching a bond valence sum close to 1 (Table 4). These O–H⋅⋅⋅Cl bonds provide further linking between Kagomé and triangular planes.
Parahibbingite is isostructural with herbertsmithite, Cu3Zn(OH)6Cl2 (Braithwaite et al., Reference Braithwaite, Mereiter, Paar and Clark2004), gillardite, Cu3Ni(OH)6Cl2 (Clissold et al., Reference Clissold, Leverett and Williams2007), tondiite, Cu3Mg(OH)6Cl2 (Malcherek et al., Reference Malcherek, Bindi, Dini, Ghiara, Molina Donoso, Nestola, Rossi and Schlüter2014) and leverettite (Kampf et al., Reference Kampf, Sciberras, Williams, Dini and Molina Donoso2013). The unit cell of these minerals is also analogous to the subcell of paratacamite [Cu2(OH)3Cl] (Welch et al., Reference Welch, Sciberras, Williams, Leverett, Schlueter and Malacherek2014). The slight differences between parahibbingite and the above-mentioned mineral structures are mainly due to the different nature of the cations at the octahedral sites.
The origin of parahibbingite crystals from the weathered Muonionalusta iron meteorite is unclear. Specimens from Bushveld Complex seem to form as a result of late hydrothermal processes: divalent iron from orthopyroxene reacted with Cl– in hydrothermal fluids creating a thin reaction rim on orthopyroxene phenocrysts. The textural relationships with other minerals suggest that parahibbingite formed after talc and amphibole, which are the earliest alteration products of the original pyroxenite (Koděra et al., Reference Koděra, Majzlan, Pollok, Kiefer, Šimko, Scholtzová, Luptáková and Cawthorn2022). In the present case, the formation process of parahibbingite associated with an iron meteorite could be similar to those involved in the alteration of archaeological iron artefacts. Similarly to the alteration products of anthropogenic objects, iron originates directly from the alloy. In this light, the iron in parahibbingite studied here most probably stems from the meteoritic Fe–Ni alloy, whereas chlorine may have been provided from the surrounding environment. Cl concentrations in the local soil are in fact remarkably high, because of the presence of significant amounts of Cl-bearing minerals, mainly scapolite- and apatite-group minerals, in the underlying bedrock (Ladenberger et al., Reference Ladenberger, Andersson, Gonzalez, Lax, Carlsson, Ohlsson and Jelinek2012; Holtstam et al., Reference Holtstam, Bindi, Karlsson, Söderhielm and Zetterqvist2020). In addition, the longtime of residence in a relatively stable and protected anoxic environment may have played an important role in the formation of large single crystals with a good degree of crystallinity. Koděra et al. (Reference Koděra, Majzlan, Pollok, Kiefer, Šimko, Scholtzová, Luptáková and Cawthorn2022) hypothesised that the β-form is actually the most stable polymorph of Fe2(OH)3Cl at ambient temperatures.
Acknowledgments
The paper benefited by the careful insights of two anonymous reviewers and the comments by the Structural Editor Peter Leverett. This research received support by Ministero dell'Istruzione, dell'Università e della Ricerca through the project PRIN 2017 “TEOREM – deciphering geological processes using Terrestrial and Extraterrestrial ORE Minerals”, prot. 2017AK8C32 (PI: Luca Bindi). Thanks are due to Anders Zetterqvist and Torbjörn Lorin for providing Muonionalusta specimens to the Swedish Museum of Natural History.
Supplementary material
To view supplementary material for this article, please visit https://doi.org/10.1180/mgm.2022.108.
Competing interests
The authors declare none.










 Open access
Open access