



Necrotising enterocolitis (NEC) is a serious cause of morbidity and mortality in premature infants, and is a major public health concern due to the increasing number of premature infants( Reference Neu and Walker 1 ). NEC is characterised by the sudden onset of ischaemia and necrosis in the small intestine typically, but can occur in the colon, affecting up to 12 % of premature infants, and has a mortality overall of up to 40 %( Reference Neu and Walker 1 – Reference Lin and Stoll 3 ), in part because current treatment options are limited to gut rest, broad-spectrum antibiotics and surgical resection of necrotic bowel( Reference Obladen 4 ). NEC is a gastrointestinal disease in its early stages, but quickly becomes a systemic disease, manifest by cardiorespiratory collapse and signs of systemic sepsis( Reference Neu and Walker 1 ). Although the precise causes of NEC remain incompletely understood, our current understanding of the disease identifies a critical link between the administration of enteral formula and its development, as demonstrated by the finding that infants who are fed breast milk are largely protected from NEC development( Reference Sullivan, Schanler and Kim 5 – Reference Meinzen-Derr, Poindexter and Wrage 8 ).
In seeking to understand the mechanisms leading to NEC development, we( Reference Leaphart, Cavallo and Gribar 9 – Reference Good, Sodhi and Egan 12 ) as well as others( Reference Jilling, Simon and Lu 13 ) have identified a critical role of the lipopolysaccharide (LPS) receptor toll-like receptor 4 (TLR4) in its pathogenesis. In a series of studies summarised in this recent review( Reference Nino, Sodhi and Hackam 14 ), we and others have shown that TLR4 activation in the intestinal epithelium leads to the loss of enterocytes through apoptosis( Reference Good, Sodhi and Egan 12 , Reference Afrazi, Branca and Sodhi 15 , Reference Egan, Sodhi and Good 16 ), followed by delayed repair through inhibition of migration and TLR4-mediated loss of intestinal stem cells( Reference Neal, Sodhi and Jia 17 ). These factors lead to the translocation of bacteria and LPS into the circulation where endothelial TLR4 activation occurs, resulting in a loss of endothelial nitric oxide synthase (eNOS), which leads to impaired perfusion and the development of experimental NEC( Reference Yazji, Sodhi and Lee 18 ).
Several groups have sought to identify potential components of breast milk that could be responsible for exerting the protection against NEC in premature infants( Reference Good, Sodhi and Egan 12 , Reference Yazji, Sodhi and Lee 18 – Reference Radulescu, Zorko and Yu 22 ), and many of these studies have shed light on the beneficial activities of human milk oligosaccharides (HMO)( Reference Newburg 23 – Reference Marcobal, Barboza and Froehlich 30 ). HMO are complex glycans that are abundant in human breast milk and not present in infant formula( Reference Newburg 23 ) and have beneficial effects on the intestine. There are various pathways by which HMO exert their protective effects on the gut, including selective consumption by protective gut microbes( Reference Wang, Li and Wu 28 ), as well as by binding to and thus preventing the adherence of bacteria to the intestinal epithelium( Reference Ruiz-Palacios, Cervantes and Ramos 31 ). HMO have also been shown to be absorbed into the circulation and excreted into the urine, suggesting that these molecules could also exert systemic effects( Reference Goehring, Kennedy and Prieto 32 , Reference Ruhaak, Stroble and Underwood 33 ), and thus may play a protective role against a disease such as NEC, which has both intestinal and systemic components( Reference Neu and Walker 1 ). Importantly, there has been no mechanism that links the potential benefits of HMO against NEC – a disease with largely systemic manifestations and not reliably linked to a particular microbe.
In the current study, we seek to evaluate the role – if any – of an abundant HMO found in breast milk – namely, 2′-fucosyllactose (HMO-2′FL) – in the protection against experimental NEC. To do so, we now focus on our recent observation that TLR4 plays a critical role in NEC development through its ability to modulate intestinal perfusion via eNOS expression as described above( Reference Yazji, Sodhi and Lee 18 ). We specifically hypothesise that administration of HMO-2′FL will reduce the incidence and severity of NEC in newborn mice through effects on mesenteric perfusion via eNOS regulation.
Methods
Statement of ethics
The described animal experiments in this study were carried out in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. Animal protocols were approved by the University of Pittsburgh’s Animal Care and Use Committee (protocol 12040382) and Johns Hopkins University (protocol M014M362).
Cells, materials, mice and reagents
LPS (Escherichia coli 0111:B4 purified by gel filtration chromatography, >99 % pure) was obtained from Sigma-Aldrich. eNOS inhibitors – N5-(1-iminoethyl)-l-ornithine dihydrochloride (L-NIO) and diphenyleneiodonium chloride (DPI) – were obtained from Santa Cruz Biotechnology. The following antibodies were obtained: eNOS (Enzo Life Sciences) and platelet endothelial cell adhesion molecule 1 (PECAM-1; BD Biosciences). The fluorescein-labelled Lycopersicon esculentum (tomato) lectin (Vector Laboratories) was used as a tracer of intestinal perfusion( Reference Yazji, Sodhi and Lee 18 ). C57BL/6 and eNOS −/− (B6·129P2-Nos3tm1Unc/J) mice were obtained from the Jackson Laboratory.
HMO-2′FL was produced through a proprietary fermentation process (Kyowa Hakko Bio Co. Ltd). Purity (95·3 %) was established by high-performance ion chromatography with pulsed amperometric detection using relative peak area comparisons. Endotoxin level (0·375 EU/mg) was estimated by limulus assay (limulus amebocyte lysate QCL-1000; Lonza).
The cell line HUV-EC-C (HUVEC) (ATCC® CRL-1730™) was obtained from American Type Culture Collection (ATCC) and maintained in endothelial cell basal medium (Cell Applications, Inc.) with low serum growth supplement (Thermo Fisher) at 37°C with 5 % CO2. Where indicated, cells were treated with HMO-2′FL (dose 100 µg/ml) 1 h before LPS administration (dose 1 µg/ml), and the induction of TLR4-mediated pro-inflammatory cytokines and eNOS expression were measured by quantitative real-time PCR (qRT-PCR) 6 h after administration of LPS. Immunohistochemistry and immunofluorescence were performed as described by Afrazi et al.( Reference Afrazi, Sodhi and Good 34 ) and evaluated using a Nikon AZ-C2+ confocal microscope (Nikon Instruments). SDS-PAGE and qRT-PCR were performed as described by Good et al.( Reference Good, Sodhi and Egan 12 ).
Induction of necrotising enterocolitis in neonatal mice
All animal experiments were approved by the University of Pittsburgh Animal Care and Use Committee. Experimental NEC was induced in 7–10-d-old mice as previously described( Reference Sodhi, Neal and Siggers 10 , Reference Afrazi, Sodhi and Good 34 ). In brief, we used formula gavage (Similac Advance infant formula (Abbott Nutrition):Esbilac (PetAg) canine milk replacer in a 2:1 ratio at a dose of 50 µl NEC formula/g body weight) five times/d, which was supplemented with enteric bacteria obtained from an infant with severe NEC as described in Good et al.( Reference Good, Sodhi and Ozolek 35 ) to simulate the dysbiosis seen in human NEC. As both the infant formula and the puppy milk replacer are commercially available, a brief description of each component of the NEC formula is as follows. Similac Advance contains the following ingredients: water, non-fat milk, lactose, high oleic safflower oil, soya oil, coconut oil, galactooligosaccharides and whey protein concentrate. Less than 0·5 % of C. cohnii oil, M. Alpina oil, β-carotene, lutein, lycopene, ascorbic acid, soya lecithin, monoglycerides, potassium citrate, calcium carbonate, potassium chloride, carrageenan, ferrous sulphate, magnesium chloride, choline chloride, choline bitartrate, taurine, m-inositol, calcium phosphate, zinc sulphate, potassium phosphate, d-α-tocopheryl acetate, niacinamide, calcium pantothenate, l-carnitine, vitamin A palmitate, cupric sulphate, thiamine chloride hydrochloride, riboflavin, pyridoxine hydrochloride, folic acid, manganese sulphate, phylloquinone, biotin, sodium selenate, vitamin D3, cyanocobalamin, salt, potassium hydroxide and nucleotides (AMP, cytidine 5′-monophosphate, disodium GMP, disodium uridine 5′-monophosphate). Esbilac canine milk replacer contains water, condensed, skimmed milk, soyabean oil, sodium caseinate, cream, calcium caseinate, l-methionine, l-arginine, calcium carbonate, choline chloride, lecithin, magnesium sulphate, potassium chloride, monopotassium phosphate, salt, tricalcium phosphate, carrageenan, dipotassium phosphate, dicalcium phosphate, ascorbic acid, ferrous sulphate, sodium hydroxide, zinc sulphate, vitamin A supplement, vitamin E supplement, niacin supplement, calcium pantothenate, copper sulphate, maltodextrins, thiamine hydrochloride, pyridoxine hydrochloride, riboflavin, manganese sulphate, vitamin D3 supplement, potassium citrate, potassium iodide, folic acid, vitamin B12 supplement and biotin.
In addition to the formula used to induce NEC, mice were also subjected to 10 min of hypoxia (5 % O2, 95 % N2) via a chamber (Billups-Rothenberg) twice a day for 4 d. Where indicated, mice were supplemented with HMO-2′FL (5 mg/ml of formula, 0·25 mg/g body weight, once daily). The eNOS inhibitor DPI was dissolved in water–dimethyl sulfoxide (DMSO) (10:90), and a 25-mm stock was prepared of which 2 µl was added to each ml of NEC formula with each feeding as indicated. A 50-mm stock of L-NIO was made with water and 1 µl was added to each ml of NEC formula with each feeding.
Necrotising enterocolitis severity assessment
Mouse terminal ileal sections were assessed by histology for the degree of mucosal injury according to our previously published scoring system from 0 (normal) to 3 (severe injury)( Reference Anand, Leaphart and Mollen 36 ), gross morphology, weight loss and by the expressions of pro-inflammatory cytokines by qRT-PCR.
Assessment of intestinal perfusion in neonatal mice
In mice in which experimental NEC was induced, intestinal perfusion was assessed as previously described in the study by Yazji et al. ( Reference Yazji, Sodhi and Lee 18 ). In brief, neonatal mice were intra-cardiacally injected with the fluorescently labelled tomato lectin (1 mg/ml, dose 5 µl/g body weight) for 5 min before euthanasia and intestinal harvest. Terminal ilea were co-stained with PECAM-1, an endothelial cell marker, and whole mounts were evaluated for PECAM-1 and tomato lectin fluorescent emission via confocal microscopy (Nikon AZ-C2+ confocal microscope). The quantification of the perfusion (villous perfusion index) was assessed as done previously( Reference Yazji, Sodhi and Lee 18 ). In brief, the sum total of tomato lectin fluorescence divided by the total volume of the mucosal vasculature is expressed as a percentage.
Measurement of nitric oxide within the intestine
Nitric oxide was measured within the intestine of mice in the indicated treatment groups using the OxiSelect™ In Vitro Nitric Oxide (Nitrite/Nitrate) Assay Kit (Cell Biolabs, Inc.) according to the manufacturer’s instructions.
16S ribosomal RNA amplicon sequence analysis
Raw paired-end reads output by the MiSeq platform was merged into consensus fragments by FLASH( Reference Magoc and Salzberg 37 ) and subsequently filtered for quality (maximum error rate 1 %) and length (minimum 200 bp) using Trimmomatic( Reference Bolger, Lohse and Usadel 38 ) and Quantitative Insights into Microbial Ecology (QIIME)( Reference Kuczynski, Stombaugh and Walters 39 , Reference Caporaso, Bittinger and Bushman 40 ). Spurious hits to the PhiX control genome were identified using Nucleotide Basic Local Alignment Search Tool (BLASTN) and removed. Passing sequences were trimmed of primers, evaluated for chimeras with UCLUST (de novo mode)( Reference Edgar, Haas and Clemente 41 ) and screened for mouse-associated contaminants using Bowtie2( Reference Langmead and Salzberg 42 ) followed by a more sensitive BLASTN search against the GreenGenes 16S database( Reference McDonald, Price and Goodrich 43 ). Chloroplast and mitochondrial contaminants were detected and filtered using the Ribosomal Database Project classifier( Reference Wang, Garrity and Tiedje 44 ) with a confidence threshold of 80 %. High-quality 16S sequences were assigned to operational taxonomic units (OTU) with a taxonomic lineage using Resphera Insight. To remove common bacterial contaminant species associated with DNA extraction kits, we first identified two well-known contaminant species present in all samples (Variovorax paradoxus and Janthinobacterium lividum) and computed Pearson’s ρ coefficient between these two species and all other OTU. Those OTU with Pearson’s ρ>0·2 for both species were removed. We further removed low-abundance contaminants on the basis of taxa identified by Salter et al.( Reference Salter, Cox and Turek 45 ). To normalise across samples, 16S profiles were subsampled to 1124 sequences per sample before downstream statistical comparisons. Differentially abundant taxa were detected using the negative binomial test with P-value correction using the false discovery rate( Reference Benjamini, Drai and Elmer 46 , Reference McMurdie and Holmes 47 ). β-Diversity calculations and principal coordinate analysis (PCoA) was performed using QIIME( Reference Kuczynski, Stombaugh and Walters 39 , Reference Caporaso, Bittinger and Bushman 40 ). Differences in β-diversity were evaluated using the Mann–Whitney U test.
Statistical analysis
In our experimental NEC experiments, each mouse represents an individual symbol on a graph. Because of different numbers of pups randomised to each group, the numbers may vary between data sets, but each is shown in the relevant data set, and statistical analysis was performed using ANOVA for multiple groups or Student’s t test for paired groups using PRISM version 6.0 (GraphPad). Statistical significance was accepted at P<0·05.
Quantitative real-time PCR
Quantitative real-time PCR was performed as previously described using the Bio-Rad CFX96 Real-Time System (Bio-Rad)( Reference Anand, Leaphart and Mollen 36 ) using the primers listed in Table 1 relative to the housekeeping gene ribosomal protein large, P0 (RPL0).
Table 1 List of primers
RPL0, ribosomal protein large, P0; TLR4, toll-like receptor 4; iNOS, inducible nitric oxide synthase; eNOS, endothelial nitric oxide synthase.
Results
Necrotising enterocolitis severity in mice is reduced by the addition of the human milk oligosaccharide 2′-fucosyllactose to the formula
As shown in Fig. 1, animals subjected to this experimental model developed gross evidence of small intestinal ischaemia, pneumatosis intestinalis and inflammation (Fig. 1(a)), as well as histological evidence of mucosal destruction and inflammatory influx (Fig. 1(b)), compared with breast-fed controls. Importantly, the administration of HMO-2′FL (0·25 mg/g body weight once daily during the model) significantly reduced the severity of NEC as compared with mice that were administered standard NEC formula, as was determined by assessment of the degree of gross and microscopic intestinal mucosal injury (Fig. 1(a) and (b)), preservation of daily weights (Fig. 1(c)), mucosal severity score (Fig. 1(d)) and by the expressions of the pro-inflammatory mediators that our laboratory( Reference Leaphart, Cavallo and Gribar 9 – Reference Good, Siggers and Sodhi 11 , Reference Egan, Sodhi and Good 16 ) and others( Reference Jain, Baggerman and Mohankumar 48 ) have shown to be important in NEC, including inducible nitric oxide synthase (iNOS), IL-6, IL-1β and TLR4 (Fig. 1(e)). Taken together, the supplementation of HMO-2′FL in the formula protects against the development of experimental NEC.

Fig. 1 The addition of the human milk oligosaccharide (HMO) 2′-fucosyllactose to infant formula attenuates necrotising enterocolitis (NEC) severity in newborn mice. (a) Representative gross images of the intestine from wild-type neonatal mice that were either breast-fed (BF) or induced to develop NEC in the absence or presence of HMO in their feeds. (b) Representative haematoxylin–eosin micrographs of the terminal ileum of wild-type mice that were either BF, breast-fed with daily administration of HMO (BF+HMO) or received NEC formula with or without the addition of HMO to their feeds (FF or FF+HMO). (c) Daily body weights (g) of experimental animals for the duration of the NEC model. ![]() , BF;
, BF; ![]() , FF;
, FF; ![]() , BF+HMO;
, BF+HMO; ![]() , FF+HMO. (d) NEC severity score (0–3) assigned by a pathologist blinded to the study conditions and treatment groups. (e) The ratio of the measured mRNA expression of the pro-inflammatory genes within the intestine including inducible nitric oxide synthase (iNOS), IL-6, IL-1
β and toll-like receptor 4 (TLR4) of the indicated groups relative to the housekeeping gene RPLO, as in the study by Good et al.(
Reference Good, Sodhi and Egan
12
). * P<0·05 v. BF control animals, ** P<0·05 v. FF. Values are means with their standard errors. Scale bar is 50 μm. Representative of three separate experiments, where each mouse represents an individual symbol on a graph. qRT-PCR, quantitative real-time PCR.
, FF+HMO. (d) NEC severity score (0–3) assigned by a pathologist blinded to the study conditions and treatment groups. (e) The ratio of the measured mRNA expression of the pro-inflammatory genes within the intestine including inducible nitric oxide synthase (iNOS), IL-6, IL-1
β and toll-like receptor 4 (TLR4) of the indicated groups relative to the housekeeping gene RPLO, as in the study by Good et al.(
Reference Good, Sodhi and Egan
12
). * P<0·05 v. BF control animals, ** P<0·05 v. FF. Values are means with their standard errors. Scale bar is 50 μm. Representative of three separate experiments, where each mouse represents an individual symbol on a graph. qRT-PCR, quantitative real-time PCR.
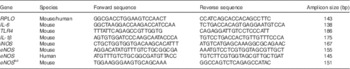
Given the possibility that there could be microbial effects, we next sought to describe the microbiota of the animals treated with HMO-2′FL. As shown in Fig. 2 and 3, taxonomic profiles at the family level reveal several well-represented taxa including Enterobacteriaceae, Lactobacillaceae and Clostridiaceae. We found Enterobacteriaceae to be generally more abundant in the experimental NEC group, whereas Lactobacillaceae appeared to be more abundant in the breast-fed group. Furthermore, differential abundance analysis between the mouse groups confirmed the associations with Enterobacteriaceae and Lactobacillaceae between the breast-fed controls as compared with animals that underwent experimental NEC; however, we did not see an association with HMO treatment (Fig. 2(b)). For Ruminococcaceae and Enterococcaceae, we observed some significant differences between HMO and their respective control groups (Fig. 2(b)). For example, Enterococcaceae was significantly more abundant in the breast-fed control group relative to the HMO-2′FL-treated group (Fig. 2(b)). We next wanted to analyse the microbial community diversity between the different treatment groups. β-Diversity analysis enabled us to compare total community composition among samples to look for broader associations beyond any single taxon. Using the Hellinger distance metric in QIIME, followed by PCoA, we observed a strong association with breast-fed compared with formula-fed (FF) animals (Fig. 3). Moreover, we can compare the β-diversity distances within and between the groups to see whether there are distinctive associations. In this case, we found that for both HMO-fed groups, the intestinal microbiomes of the mice were significantly more homogeneous than the breast-fed and FF groups (Fig. 3).
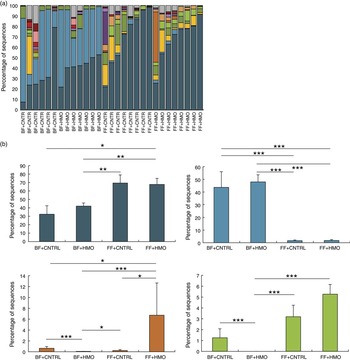
Fig. 2 Characterisation of the microbiota in mice treated with the human milk oligosaccharide (HMO) 2′-fucosyllactose (2′FL). (a) Taxonomic profiles at the family level for the microbiotas of pups treated with HMO-2′FL with necrotising enterocolitis (NEC) or breast-fed controls. ![]() , Other taxa;
, Other taxa; ![]() , Proteobacteria_Alphaproteobacteria_Sphingomonadales_Sphingomonadaceae;
, Proteobacteria_Alphaproteobacteria_Sphingomonadales_Sphingomonadaceae; ![]() , Proteobacteria_Gammaproteobacteria_Pseudomonadales_Moraxellaceae;
, Proteobacteria_Gammaproteobacteria_Pseudomonadales_Moraxellaceae; ![]() , Proteobacteria_Gammaproteobacteria_Pseudomonadales_Pseudomonadaceae;
, Proteobacteria_Gammaproteobacteria_Pseudomonadales_Pseudomonadaceae; ![]() , Firmicutes_Bacilli_Bacillales_Staphylococcaceae;
, Firmicutes_Bacilli_Bacillales_Staphylococcaceae; ![]() , Proteobacteria_Gammaproteobacteria_Pasteurellales_Pasteurellaceae;
, Proteobacteria_Gammaproteobacteria_Pasteurellales_Pasteurellaceae; ![]() , Firmicutes_Clostridia_Clostridiales_Incertae sedis;
, Firmicutes_Clostridia_Clostridiales_Incertae sedis; ![]() , Firmicutes_Clostridia_Clostridiales_Ruminococcaceae;
, Firmicutes_Clostridia_Clostridiales_Ruminococcaceae; ![]() , Firmicutes_Bacilli_Lactobacillales_Enterococcaceae;
, Firmicutes_Bacilli_Lactobacillales_Enterococcaceae; ![]() , Firmicutes_Bacilli_Lactobacillales_Streptococcaceae;
, Firmicutes_Bacilli_Lactobacillales_Streptococcaceae; ![]() , Firmicutes_Clostridia_Clostridiales_Clostridiaceae 1;
, Firmicutes_Clostridia_Clostridiales_Clostridiaceae 1; ![]() , Firmicutes_Bacilli_Lactobacillales_Lactobacillaceae;
, Firmicutes_Bacilli_Lactobacillales_Lactobacillaceae; ![]() , Proteobacteria_Gammaproteobacteria_Enterobacteriales_Enterobacteriaceae. (b) Differential abundance analysis of the indicated treatment groups.
, Proteobacteria_Gammaproteobacteria_Enterobacteriales_Enterobacteriaceae. (b) Differential abundance analysis of the indicated treatment groups. ![]() , Enterobacteriaceae;
, Enterobacteriaceae; ![]() , Lactobacillaceae;
, Lactobacillaceae; ![]() , Ruminococcaceae;
, Ruminococcaceae; ![]() , Enterococcaceae. BF, breast-fed; CNTRL, control; FF, formula fed. * Adjusted P<0·05; ** adjusted P<0·01; *** adjusted P<0·001.
, Enterococcaceae. BF, breast-fed; CNTRL, control; FF, formula fed. * Adjusted P<0·05; ** adjusted P<0·01; *** adjusted P<0·001.

Fig. 3
β-Diversity analysis of the microbiota in mice treated with the human milk oligosaccharide (HMO) 2′-fucosyllactose. (a, b) β-Diversity analysis of the microbiota of the indicated treatment groups. Differences in β-diversity were evaluated using the Mann–Whitney U test. BF, breast-fed; CNTRL, control; FF, formula fed. * P<0·05; ** P<0·01; *** P<0·001. ![]() , BF+CNTRL;
, BF+CNTRL; ![]() , BF+HMO;
, BF+HMO; ![]() , FF+CNTRL;
, FF+CNTRL; ![]() , FF+HMO. PC1, principal component 1; PC2, principal component 2.
, FF+HMO. PC1, principal component 1; PC2, principal component 2.
The addition of the human milk oligosaccharide 2′-fucosyllactose prevented the inhibition on mesenteric perfusion observed in experimental necrotising enterocolitis in newborn mice
In order to define the mechanisms by which HMO-2′FL could protect against the development of NEC, we next turned our attention to the effects of HMO-2′FL on the potential regulation of eNOS-mediated mesenteric blood flow by performing mesenteric micro-angiography in mice that underwent experimental NEC. To do so, we first assessed the degree of mesenteric perfusion in the mouse model of experimental NEC in the presence or absence of HMO-2′FL supplementation to the formula. As shown in Fig. 4, the intestinal perfusion in mice that were FF was significantly reduced compared with breast-fed controls, consistent with our previous findings(18). Importantly, the addition of HMO-2′FL restored perfusion to levels comparable with that of mice that were breast-fed on each day of the model, as observed in Fig. 4(a) and quantified in Fig. 4(b). The improvement in perfusion corresponded to a pink and healthy appearing intestine in breast-fed mice as compared with the dusky bowel seen in NEC, which was reversed in the presence of HMO-2′FL towards a healthy, pink and perfused appearance. Taken together, these findings illustrate that HMO-2′FL restores intestinal perfusion in NEC. We next sought to explore the potential mechanisms involved and focused on the effects of the vasodilatory molecule eNOS.
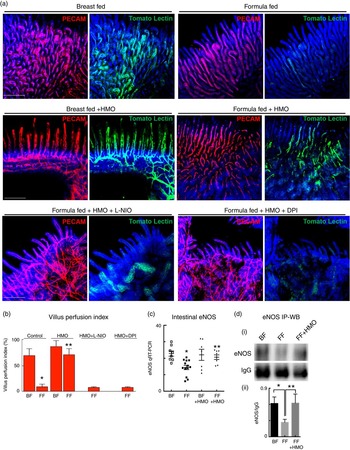
Fig. 4 Formula supplementation with the human milk oligosaccharide (HMO) 2′-fucosyllactose enhances mesenteric perfusion in experimental necrotising enterocolitis (NEC) via maintenance of intestinal endothelial nitric oxide synthase (eNOS) expression. (a) Representative confocal micrographs of terminal ileal whole mounts from wild-type neonatal mice from the indicated treatment groups: breast-fed controls (BF), breast-fed with HMO (BF+HMO, experimental NEC, formula fed (FF) and FF with HMO (FF+HMO), as well as mice that in addition to the experimental NEC model and HMO treatment also received the eNOS inhibitors N5-(1-iminoethyl)-l-ornithine dihydrochloride (L-NIO) (FF+HMO+L-NIO) or diphenyleneiodonium chloride (DPI) (FF+HMO+DPI) with their formula. After intra-cardiac injection with the fluorescent tracer tomato lectin, which correlates with intestinal blood flow (green), whole mounts were immunostained for platelet endothelial cell adhesion molecule 1 (PECAM-1) to assess the intestinal microvasculature (red). (b) Graph representing villus perfusion index as described in the Materials and methods section expressed as a percentage. (c) Intestinal eNOS mRNA expression by quantitative real-time PCR (qRT-PCR) in the indicated treatment groups relative to the housekeeping gene RPLO. (d) SDS-PAGE immunoprecipitation Western blot (IP-WB) performed on the terminal ileum of wild-type mice in the indicated treatment groups. IgG is shown as a loading control (i); densitometry quantification with Image J performed on three samples per group in (ii). * P<0·05 v. breast-fed control animals, ** P<0·05 v. FF. Values are means with their standard errors. Scale bar is 50 μm. Representative of at least three separate experiments, where each mouse represents an individual symbol on a graph.
Human milk oligosaccharide 2′-fucosyllactose maintains the expression of endothelial nitric oxide synthase in necrotising enterocolitis, and thus restores intestinal perfusion in neonatal mice
Having now shown that HMO-2′FL restores mesenteric perfusion in NEC, we next sought to investigate the possibility that HMO-2′FL may affect the expression of eNOS. To do so, we assessed the expression of eNOS in the intestine of mice with and without NEC in the presence or absence of HMO-2′FL. As shown in Fig. 4, the expression of eNOS in the intestine was significantly reduced in FF mice compared with breast-fed control mice, and this was restored after HMO-2′FL induction (Fig. 4(c) and (d (i, ii))). The finding that HMO-2′FL could restore the expression of eNOS in the newborn gut in the setting of NEC raises the intriguing possibility that perhaps the protection against NEC by HMO-2′FL occurs via induction of eNOS expression. To assess this directly, we next utilised two approaches to inhibit eNOS – the use of pharmacological inhibitors and the use of eNOS knockout mice – and sought to determine whether HMO-2′FL could still protect against NEC development under conditions of eNOS inhibition. As shown in Fig. 5, we next treated mice with eNOS inhibitors L-NIO dihydrochloride and DPI. Importantly, under conditions of eNOS inhibition (verification in the online Supplementary Fig. S1(a)), HMO-2′FL failed to protect against NEC as demonstrated by histology, the pro-inflammatory cytokine IL-1 β and NEC severity score (Fig. 5(a) and (c)), revealing that HMO-2′FL acts in part via eNOS activity. To assess whether eNOS expression was required for the protection exerted by HMO-2′FL against NEC, we next performed studies in eNOS −/− mice (Fig. 6, online Supplementary Fig. S1(b)) and as shown in the study by Shesely et al. ( Reference Shesely, Maeda and Kim 49 ). As shown in Fig. 6, eNOS −/− mice were significantly vulnerable to experimental NEC, consistent with our previous studies( Reference Yazji, Sodhi and Lee 18 ), and administration of HMO-2′FL did not confer protection as demonstrated by a disruption in the intestinal mucosal architecture, increase in pro-inflammatory cytokines and NEC severity, as well as significantly reduced intestinal perfusion (Fig. 6(a) and (d)). Taken together, these findings demonstrate that HMO-2′FL is protective against experimental NEC in a manner that is dependent on the expression and function of eNOS.
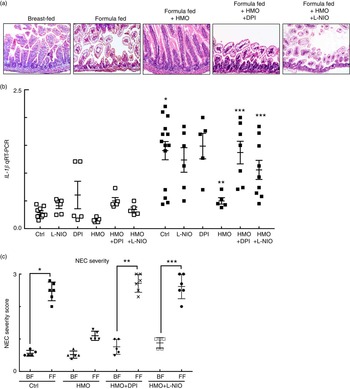
Fig. 5 The human milk oligosaccharide (HMO), 2′-fucosyllactose failed to protect against necrotising enterocolitis (NEC) in the presence of endothelial nitric oxide synthase inhibition. (a) Representative haematoxylin–eosin micrographs of the terminal ileum of wild-type mice in the indicated treatment groups: breast-fed controls (Ctrl), formula fed (FF), FF plus HMO (FF+HMO) and FF+HMO plus diphenyleneiodonium chloride (DPI) (FF+HMO+DPI) or FF+HMO and N5-(1-iminoethyl)-l-ornithine dihydrochloride (L-NIO) (FF+HMO+L-NIO). (b) IL-1
β mRNA expression from the terminal ileum of the mice in the indicated treatment groups (![]() , breast-fed mice;
, breast-fed mice; ![]() , FF mice with indicated treatment groups) relative to the housekeeping gene RPLO. * P<0·05 v. breast-fed control (Ctrl) animals, ** P<0·05 v. FF control (Ctrl), *** P<0·05 v. FF+HMO. (c) NEC severity score assessed by a pathologist blinded to the study conditions. * P<0·05 v. breast-fed controls, ** P<0·05 v. breast-fed+HMO+DPI, *** P<0·05 v. breast-fed+HMO+L-NIO. Values are means with their standard errors. Scale bar is 50 μm. Representative of at least three separate experiments, where each mouse represents an individual symbol on a graph.
, FF mice with indicated treatment groups) relative to the housekeeping gene RPLO. * P<0·05 v. breast-fed control (Ctrl) animals, ** P<0·05 v. FF control (Ctrl), *** P<0·05 v. FF+HMO. (c) NEC severity score assessed by a pathologist blinded to the study conditions. * P<0·05 v. breast-fed controls, ** P<0·05 v. breast-fed+HMO+DPI, *** P<0·05 v. breast-fed+HMO+L-NIO. Values are means with their standard errors. Scale bar is 50 μm. Representative of at least three separate experiments, where each mouse represents an individual symbol on a graph.
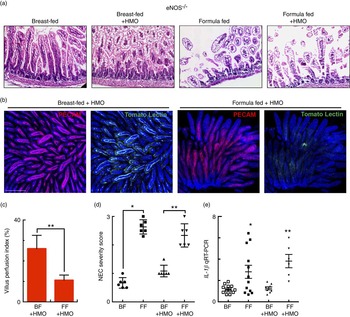
Fig. 6 The human milk oligosaccharide (HMO) 2′-fucosyllactose does not protect against necrotising enterocolitis (NEC) or enhance intestinal perfusion in endothelial nitric oxide synthase (eNOS)−/− mice. (a) Representative haematoxylin–eosin micrographs of the terminal ileum of eNOS −/− mice in the indicated treatment groups. (b) Representative confocal micrographs of terminal ileal whole mounts from eNOS −/− mice that received HMO and were either breast-fed (BF+HMO) or were formula-fed (FF+HMO). Whole mounts were stained for platelet endothelial cell adhesion molecule 1 (PECAM-1) to assess the microvasculature of the intestine and subjected to intra-cardiac injections with tomato lectin as a marker of intestinal perfusion (green). (c) Graph representing villus perfusion index as described in the Materials and methods section expressed as a percentage. (d) NEC severity score assessed by a pathologist blinded to the study conditions. (e) IL-1 β mRNA expression from the terminal ileum of the mice in the indicated treatment groups. * P<0·05 v. breast-fed control animals, ** P<0·05 v. BF+HMO. Values are means with their standard errors. Scale bar is 50 μm. Representative of at least three separate experiments, where each mouse represents an individual symbol on a graph. qRT-PCR, quantitative real-time PCR.
Lipopolysaccharide decreases the expression of endothelial nitric oxide synthase in HUVEC endothelial cells and this decrease is attenuated by human milk oligosaccharide 2′-fucosyllactose
In the final series of studies, we sought to explore in greater detail the potential mechanisms by which HMO-2′FL could regulate eNOS expression – and therefore mesenteric perfusion – in the newborn gut. To do so, we first examined whether HMO-2′FL could regulate the mRNA expression of eNOS in HUVEC endothelial cells in the presence of either LPS as a TLR4 ligand or LPS in the presence of HMO-2′FL to mimic the situation observed in the mesenteric endothelium. As shown in Fig. 7, TLR4 activation reduced the expression of eNOS mRNA in HUVEC (Fig. 7(a)), consistent with our previous findings in primary cultured endothelial cells( Reference Yazji, Sodhi and Lee 18 ). Importantly, the addition of HMO-2′FL restored eNOS expression as demonstrated by immunocytochemistry (Fig. 7(b)), consistent with the intestinal protein expression shown in Fig. 4(d), and demonstrated a clear role of HMO-2′FL in the regulation of eNOS gene expression and function.

Fig. 7 Expression of endothelial nitric oxide synthase (eNOS) is enhanced by the human milk oligosaccharide (HMO) 2′-fucosyllactose in HUV-EC-C (HUVEC). (a) eNOS mRNA by quantitative real-time PCR (qRT-PCR) from HUVEC in the indicated treatment groups: vehicle control (Ctrl), lipopolysaccharide (LPS), HMO alone (HMO) and LPS with HMO (LPS+HMO) relative to the housekeeping gene RPLO. (b) Representative confocal micrographs of HUVEC with the indicated treatment groups stained for eNOS (red) and 4,6-diamidino-2-phenylindole, dihydrochloride (DAPI) (blue). * P<0·05 v. vehicle control, ** P<0·05 v. LPS. Values are means with their standard errors. Scale bar is 10 μm. Results representative of three separate experiments with over 50 high power fields per group imaged.
Discussion
In the current study, we utilised an experimental mouse model of NEC, which recapitulates several of the features seen in premature infants with NEC, including gross evidence of small intestinal ischaemia, pneumatosis intestinalis and inflammation, as well as histological evidence of mucosal destruction( Reference Good, Sodhi and Egan 12 , Reference Afrazi, Branca and Sodhi 15 , Reference Egan, Sodhi and Good 16 , Reference Yazji, Sodhi and Lee 18 ), to demonstrate that the administration of formula containing an abundant HMO in human milk – namely, 2′FL – significantly reduces the severity of experimental NEC in newborn mice. In seeking to define the mechanisms involved, we and others have shown that TLR4 signalling in the gut leads to NEC via mucosal disruption, leading to LPS translocation and a loss of eNOS, which results in impaired perfusion and the development of intestinal ischaemia( Reference Yazji, Sodhi and Lee 18 , Reference Yu, Radulescu and Zorko 50 ). We have now determined that HMO-2′FL-supplemented formula acts by regulating the degree of blood flow to the newborn intestine via eNOS, and that this novel pathway may explain some of the previously unrecognised benefits of this breast milk oligosaccharide.
The current findings may shed light on the impact of other aspects of maternal breast milk production that could impact on the likelihood of NEC development to occur. Specifically, previous authors have noted that nearly 20 % of women in the population are homozygous for common mutations in the secretor gene fucosyltransferase 2 (FUT2) and are unable to produce HMO-2′FL( Reference Demmert, Schaper and Pagel 51 ), which has been shown to impact the intestinal microbiota of their infants( Reference Wacklin, Makivuokko and Alakulppi 52 ). The studies on the relationship of FUT2 mutations and risk for NEC to date have focused predominantly on the phenotype or genotype of the baby rather than the mother( Reference Demmert, Schaper and Pagel 51 , Reference Wacklin, Makivuokko and Alakulppi 52 ). We now speculate that any increase in NEC in premature infants receiving milk from homozygous FUT2 mutant mothers may reflect a lack of endogenous 2′FL, suggesting that supplementing their milk with 2′FL could benefit their premature infants through NEC protection.
Our findings extend our knowledge on the mechanisms by which HMO act on the neonatal intestine, and provide additional insights into the benefit of breast milk that is observed in premature infants at risk for developing NEC. Specifically, the molecular details underlying the capacity of two genera of gut bacteria to consume HMO have been demonstrated, including Bifidobacterium( Reference Garrido, Barile and Mills 53 ) and Bacteroidetes( Reference Marcobal and Sonnenburg 54 ). Indeed, the presence of gram-negative bacteria including E. coli and Klebsiella has been associated with NEC outbreaks, and are commonly seen in the blood and stool of patients with this disease( Reference Kosloske and Ulrich 55 ). HMO have also been shown to have structural homology to cell surface glycans, and can therefore act as molecular mimics, or decoys, by binding to luminal bacteria, and thereby prevent binding to the intestinal epithelium( Reference Morrow, Ruiz-Palacios and Jiang 56 ), thus potentially reducing the degree of bacterial inflammation and activation of toll-like receptors on the intestinal epithelium, which we have shown to be required for NEC development( Reference Sodhi, Neal and Siggers 10 , Reference Afrazi, Branca and Sodhi 15 , Reference Neal, Sodhi and Jia 17 , Reference Afrazi, Sodhi and Good 34 , Reference Anand, Leaphart and Mollen 36 , Reference Gribar, Sodhi and Richardson 57 ). Moreover, analysis of the microbiota revealed that supplementation of HMO-2′FL to the experimental NEC formula increased the faecal content of Ruminococcaceae, a family of bacteria of class Clostridia, which typically increase in premature infants as they mature and approach 36 weeks of postconceptional age( Reference La Rosa, Warner and Zhou 58 ). Although HMO-2′FL appears to affect the microbiota, these changes do not seem to be the primary mechanism of protection against NEC. The current findings extend the studies by Goehring et al.( Reference Goehring, Kennedy and Prieto 32 ) who demonstrated that in addition to these local effects HMO are absorbed systemically in human infants, which in this case explains the effect on mesenteric perfusion through the activity of the vasodilator eNOS. Furthermore, 2′FL has been demonstrated in the plasma of breast-fed infants, although in lesser abundance than several other HMO( Reference Ruhaak, Stroble and Underwood 33 ), and in the urine of breast milk-fed premature infants( Reference Underwood, Gaerlan and De Leoz 59 ). Although our studies did not directly test the systemic absorption of 2′FL, other authors have elegantly shown that HMO are absorbed in the intestine and can be measured in the serum and urine of rats, demonstrating the significance of the effects of HMO on other species( Reference Jantscher-Krenn, Zherebtsov and Nissan 19 , Reference Santos-Fandila, Zafra-Gomez and Vazquez 60 ). In determining which HMO are present in mouse breast milk, Prieto et al.( Reference Prieto, Mukerji and Kelder 61 ) have shown that 2′FL is not present in mouse milk, while 3’FL indeed is. Importantly, the biological significance of the differential expression of HMO in breast milk across various species is unknown. It is noteworthy that the current study is supported by previous studies by Jantscher-Krenn et al.( Reference Jantscher-Krenn, Zherebtsov and Nissan 19 ) who showed that the addition of the HMO disialyllacto-N-tetraose prevents NEC in neonatal rats. Although these studies did not provide a mechanism to explain these protective effects, and indeed the effects may be structure specific, we now speculate that the protection achieved was similar to that observed in the current study, and involved an enhanced mesenteric perfusion. Additional effects on the microbiota and on bacterial enterocyte signalling may also play a role in the protection in these two studies, and additional work will be required to determine these precise effects. It is possible that 2′FL gets absorbed into the circulation, attenuates endothelial TLR4 signalling and subsequently maintains eNOS levels. Moreover, it is likely that breast milk has various direct TLR4 inhibitors, as we have recently identified a class of small molecule inhibitors of TLR4( Reference Neal, Jia and Eyer 62 ), which are themselves oligosaccharides, suggesting that HMO-2′FL itself may inhibit TLR4 signalling. In support of this possibility, the expression of TLR4 was reduced in the mucosa of mice after administration of HMO-2′FL. Furthermore, it has recently been reported that HMO-2′FL modulates the expression of cluster of differentiation 14 (CD14), a molecule that is essential for LPS binding and optimal TLR4 signalling( Reference He, Liu and Kling 63 ). The current study thus advances our understanding of the mechanisms by which breast milk may protect against the development of NEC.
In summary, the current findings reveal an important role for HMO-2′FL in the protection against the development of NEC in newborn mice through the maintenance of mesenteric perfusion via a mechanism that requires the expression of eNOS. These findings expand our understanding about the protective benefits of breast milk for premature infants, in which HMO are present in high amounts, and also indicate that in addition to the established roles of HMO on the microbiota or on the epithelial surface, these molecules may have key roles when absorbed systemically. Although our preclinical studies were performed in neonatal mice rather than premature infants, the current studies also suggest that 2′FL is one of the components within human breast milk found to be protective against NEC, which remains an important goal of neonatal nutrition research. Furthermore, these studies reinforce the statements by the American Academy of Pediatrics Section on Breastfeeding and the European Society for Pediatric Gastroenterology, Hepatology and Nutrition that the preferred feeding for premature infants should be the mother’s own milk with donor human milk from an established human milk bank as a second choice if the mother’s milk supply is inadequate( Reference Eidelman, Schanler and Johnston 64 , Reference Nutrition, Arslanoglu and Corpeleijn 65 ).
Acknowledgements
M. G. is supported by K08DK101608 from the National Institutes of Health and the Children’s Hospital of Pittsburgh of the UPMC Health System. D. J. H. is supported by a sponsored research grant from Abbott Nutrition, as well as R01DK083752 and R01GM078238 from the National Institutes of Health.
M. G., C. P. S., R. H. B., K. C. G. and D. J. H. designed and/or performed experiments, analysed results, wrote and edited the manuscript; Y. Y., H. J., P. L., W. B. F., L. Y. M., T. P., D. F. N., Q. Z. and C. M. performed experiments and analysed data.
The authors declare that there are no conflicts of interest.
Supplementary material
For supplementary material/s referred to in this article, please visit http://dx.doi.org/doi:10.1017/S0007114516002944










