


Introduction
Coronaviruses (CoVs) form a large group of enveloped viruses that harbour the largest genome among all RNA viruses (26.4–31.7 kb in length). CoVs occupy the entire subfamily of Coronavirinae within the family Coronaviridae, order Nidovirales. Traditionally, CoVs were classified into three groups depending on their antigenic and genetic properties (Brian and Baric, Reference Brian and Baric2005). However, these groups have been recently replaced with four genera: Alphacoronavirus, Betacoronavirus, Gammacoronavirus and Deltacoronavirus. The first two genera include only mammalian CoVs, while all avian CoVs are members of the other two genera (de Groot et al., Reference de Groot, Baker, Baric, Enjuanes, Gorbalenya, Holmes, Perlman, Poon, Rottier, Talbot, Woo, Ziebuhr, King, Adams, Carstens and Lefkowitz2012).
CoVs infect a wide diversity of animal and bird species causing respiratory, enteric, neurologic and hepatic disorders. Since SARS-CoV was identified in 2003, a significant increase in the number of emerging CoVs has been shown in people and animals as a result of the growing interest in CoV research and the development of improved diagnostic tools (Woo et al., Reference Woo, Lau, Huang and Yuen2009; Peck et al., Reference Peck, Burch, Heise and Baric2015). The accumulated information has confirmed the ability of CoVs to adapt new tissue tropisms, to jump host-species barriers and to acclimatize variable ecological niches (Decaro et al., Reference Decaro, Mari, Elia, Addie, Camero, Lucente, Martella and Buonavoglia2010b). These abilities were believed to be attributed to: (1) the high mutation rate (a single mutation/genome/round of replication) caused by low fidelity of the viral RNA polymerase (Drake and Holland, Reference Drake and Holland1999), and (2) the exceptional tendency for homologous recombination mediated by random template switching during RNA replication (Woo et al., Reference Woo, Lau, Yip, Huang, Tsoi, Chan and Yuen2006; Decaro and Buonavoglia, Reference Decaro and Buonavoglia2008). Cross-species transmission of CoVs was previously recorded for many CoVs [e.g. SARS-CoV in palm civet and human beings (Song et al., Reference Song, Tu, Zhang, Wang, Zheng, Lei, Chen, Gao, Zhou, Xiang, Zheng, Chern, Cheng, Pan, Xuan, Chen, Luo, Zhou, Liu, He, Qin, Li, Ren, Liang, Yu, Anderson, Wang, Xu, Wu, Zheng, Chen, Liang, Gao, Liao, Fang, Jiang, Li, Chen, Di, He, Lin, Tong, Kong, Du, Hao, Tang, Bernini, Yu, Spiga, Guo, Pan, He, Manuguerra, Fontanet, Danchin, Niccolai, Li, Wu and Zhao2005); Middle East respiratory syndrome CoV (MERS-CoV) in camels and people (Drosten et al., Reference Drosten, Kellam and Memish2014)]. Bovine CoV (BCoV) represents an excellent example of a CoV that extensively crosses the interspecies barrier. Several bovine-like CoVs were identified as aetiologic pathogens of enteric and/or respiratory diseases in a diverse spectrum of ruminant species (Saif, Reference Saif2010), dogs (Decaro et al., Reference Decaro, Desario, Elia, Mari, Lucente, Cordioli, Colaianni, Martella and Buonavoglia2007; Erles et al., Reference Erles, Shiu and Brownlie2007; Lorusso et al., Reference Lorusso, Desario, Mari, Campolo, Lorusso, Elia, Martella, Buonavoglia and Decaro2009) and even human beings (Zhang et al., Reference Zhang, Herbst, Kousoulas and Storz1994).
Bovine CoV
BCoVs are commonly identified in the respiratory and intestinal tracts of healthy cattle, and contribute to different disease syndromes (Fulton et al., Reference Fulton, Herd, Sorensen, Confer, Ritchey, Ridpath and Burge2015). The virus is involved as a major cause of calf diarrhoea (CD) during the first 3 weeks of life in both dairy and beef cattle herds (Boileau and Kapil, Reference Boileau and Kapil2010). High mortalities are mostly attributed to BCoV infections because they affect both small and large intestines, destroying villi and leading to severe, often bloody, diarrhoea (Torres-Medina et al., Reference Torres-Medina, Schlafer and Mebus1985). Enteric BCoV infection is also associated with winter dysentery (WD) in adult dairy cattle, causing dramatic decrease in milk production and significant economic losses (Natsuaki et al., Reference Natsuaki, Goto, Nakamura, Yamada, Ueo, Komori, Shirakawa and Uchinuno2007). Respiratory tract infection in growing (2–16 weeks of age) and feedlot calves has been frequently attributed to BCoV (Storz et al., Reference Storz, Purdy, Lin, Burrell, Truax, Briggs, Frank and Loan2000; Decaro et al., Reference Decaro, Mari, Desario, Campolo, Elia, Martella, Greco, Cirone, Colaianni, Cordioli and Buonavoglia2008b; Saif, Reference Saif2010). The widespread distribution of BCoV could be explained by two main factors: (1) rapid transmission of the virus via faecal–oral and respiratory routes (Heckert et al., Reference Heckert, Saif and Agnes1990; Decaro et al., Reference Decaro, Campolo, Desario, Cirone, D'Abramo, Lorusso, Greco, Mari, Colaianni, Elia, Martella and Buonavoglia2008a), and (2) existence of carrier animals within infected herds. These carriers shed the virus in faeces, particularly during the stress of winter season and parturition, and serve as a source of infection to their neonates and reservoirs for reinfection on the farm (Carman and Hazlett, Reference Carman and Hazlett1992).
Bovine-like CoVs
In 2008, the Coronavirus Study Group of the International Committee of Taxonomy of Viruses has proposed taxonomic grouping of six CoVs of people, cattle, pigs, horses and dogs [human CoV OC43 (HCoV-OC43), human enteric CoV, BCoV, porcine haemagglutinating encephalomyelitis virus, equine CoV and canine respiratory CoV] in a new virus species named Betacoronavirus 1 (de Groot et al., Reference de Groot, Ziebuhr, Poon, Woo, Talbot, Rottier, Holmes, Baric, Perlman and Enjuanes2008). Due to their close antigenic and genetic relatedness, members of Betacoronavirus 1 appear as host-range variants rather than separate viruses. It has been concluded that they may originate as a result of genetic recombination and interspecies transmission events (Zhang et al., Reference Zhang, Herbst, Kousoulas and Storz1994; Lau et al., Reference Lau, Lee, Tsang, Yip, Tse, Lee, So, Lau, Chan, Woo and Yuen2011; Lau et al., Reference Lau, Woo, Yip, Fan, Huang, Wang, Guo, Lam, Tsang, Lai, Chan, Che, Zheng and Yuen2012). For instance, HCoV-OC43 likely evolved from ancestral BCoV strains that crossed the interspecies barrier and established an infection in human beings around 1890 (Vijgen et al., Reference Vijgen, Keyaerts, Moes, Thoelen, Wollants, Lemey, Vandamme and Van Ranst2005; Vijgen et al., Reference Vijgen, Keyaerts, Lemey, Maes, Van Reeth, Nauwynck, Pensaert and Van Ranst2006).
Several CoV strains have been identified in the faeces, intestinal contents and sometimes in the respiratory secretions of a diverse group of domestic and wild (captive and free-range) ruminant species (Tables 1 and 2). Most of these viruses have demonstrated extensive biologic, antigenic and genetic similarities with BCoV and consequently they were termed as bovine-like CoVs (Tsunemitsu et al., Reference Tsunemitsu, El-Kanawati, Smith, Reed and Saif1995). Experimental transmission of bovine-like CoVs to gnotobiotic and colostrum-deprived cattle calves suggest that cattle may be reservoirs for CoV strains to infect other domestic and wild ruminants and vice versa, in a process that helps maintaince and evolution of CoVs in these species (Tsunemitsu et al., Reference Tsunemitsu, El-Kanawati, Smith, Reed and Saif1995; Hasoksuz et al., Reference Hasoksuz, Alekseev, Vlasova, Zhang, Spiro, Halpin, Wang, Ghedin and Saif2007). It has been proposed that the broad host range of BCoV may be attributed to the presence of haemagglutinin-esterase protein that enables the virus to bind to different cell types (Saif, Reference Saif2010).
Table 1. A collective summary of bovine-like CoVs in domestic ruminants

VNT, virus neutralization test; HI, haemagglutination inhibition; HEHA, haemadsorption–elution–haemagglutination assays; ND, not defined; ELISA, enzyme-linked immunosorbent assay; IP, immunoperoxidase; NC-EM, negative contrast electron microscopy; HA, haemagglutination; RT-PCR, reverse transcription polymerase chain reaction; EM, electron microscopy; IHC, immunohistochemistry; IFA, immunofluorescent assay; UAE, United Arab of Emirates; IC, immunochromatography.
Table 2. A collective summary of bovine-like CoVs in wild ruminants
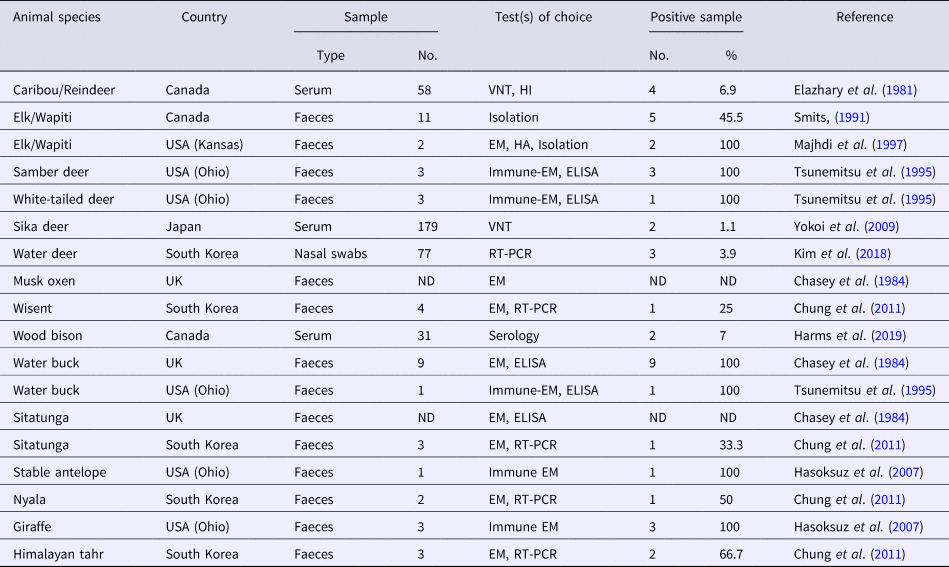
VNT, virus neutralization test; HI, haemagglutination inhibition; EM, electron microscopy; HA, haemagglutination; ELISA, enzyme-linked immunosorbent assay; ND, not defined; RT-PCR, reverse transcription polymerase chain reaction.
Bovine-like CoVs in domestic ruminants
Water buffalo (Bubalus bubalis)
The domestic Asian buffalo (commonly named as water buffalo) is a large bovid originated from Southeastern Asia and tamed approximately 5000 years ago. It is used in many countries as a source of good quality meat, milk and leather, and as a labour animal in agriculture farms. The population of water buffalo in the world was estimated to be 172 million head with 95% living in Asia (mostly in India, China and Pakistan), 2.4% in South America (principally in Brazil), 2.3% in Africa (only in Egypt), 0.3% in Europe and 0.02% in Australia (Borghese, Reference Borghese2011).
The first description of bovine-like CoVs in water buffalo dated back to 1985, when BCoV antibodies were detected using virus neutralization (VN) and haemagglutination inhibition (HI) assays in 52.9% of sera collected from water buffaloes of different age groups in Bulgaria. Seven faecal samples of diarrhoeic buffalo calves showed haemagglutinating activity that was suppressed by BCoV-specific antiserum (Muniiappa et al., Reference Muniiappa, Mitov and Kharalambiev1985). In the early 1990s, bovine-like CoVs were detected in the faeces of suckling buffalo calves with profuse diarrhoea in Egypt using monoclonal antibody (MAb)-based enzyme-linked immunosorbent assay (ELISA) and immunoperoxidase assays (Abd El-Karim et al., Reference Abd El-Karim, El-Sanousi, Aly and Reda1990). The CoV particles were identified in two further studies in Egypt using negative-contrast transmission electron microscopy (EM) in 20 and 55.2% of the faecal samples collected from diarrhoeic buffalo calves, and 8.3 and 19.1% of the faecal samples collected from apparently healthy buffalo calves, respectively (Byomi et al., Reference Byomi, Herbst and Baljer1996; Abd El-Rahim, Reference Abd El-Rahim1997). The epidemiological data presented by the Tri-national Health Research Project, USAID, indicated that CoV is the second most-common pathogen causing diarrhoea in buffalo calves in Egypt with a prevalence rate of 37.7% (Saleh, Reference Saleh1994; Garbe et al., Reference Garbe, Amarin and Hicheri1995).
An outbreak of CD and neonatal mortality was later described in a herd of water buffaloes in Campania (Southern Italy) between October 2006 and April 2007. Bovine-like CoVs were identified in the intestinal contents of buffalo calves with severe diarrhoea (two dead and 17 alive) by conventional and real-time reverse transcription polymerase chain reaction (RT-PCR) assays. A virus strain (designated as 179/07-11) was isolated with HRT-18 cells, producing the typical cytopathic effects of BCoV. This strain shared many similarities with BcoVs including: (1) haemagglutination of mouse erythrocytes, (2) receptor-destroying enzymatic activity, (3) cross-reactivity with BCoV antisera in immunofluorescence (IF) assay, and (4) genomic organization. However, the inability of strain 179/07-11 to replicate in MDBK cells and to agglutinate chicken erythrocytes, along with the existence of considerable genetic distance between both bovine and buffalo strains, suggested the assignment of strain 179/07-11 as a prototype of a novel host-range variant of BCoV (named as bubaline CoV) (Decaro et al., Reference Decaro, Martella, Elia, Campolo, Mari, Desario, Lucente, Lorusso, Greco, Corrente, Tempesta and Buonavoglia2008c). Four further bubaline CoV strains were isolated afterwards and all showed similar biologic and antigenic characteristics to the prototype strain 179/07-11. Nevertheless, the genetic analysis showed that three of these strains were closely related to the prototype strain whereas the fourth shared higher genetic similarity to recent BCoV reference strains (Decaro et al., Reference Decaro, Cirone, Mari, Nava, Tinelli, Elia, Sarno, Martella, Colaianni, Aprea, Tempesta and Buonavoglia2010a). It is proposed that the bubaline CoV may be originated from interspecies transmission of a BCoV strain; however, no cattle, sheep or goat contact to the water buffalo herd was demonstrated at the time of the outbreak.
The first complete genome sequence of a bubaline CoV was retrieved from two strains identified in Bangladesh in 2014 (BufCoV HKU26 strains B1-24F and B1-28F). The overall nucleotide homology with BCoVs was ranged from 98 to 99%, which supports the taxonomic relevance of both types of CoVs. Nevertheless, the nature of the three accessory proteins that located between spike (S) and envelop (E) varies distinctively. The 4.9 kDa protein of BCoV was replaced with 2.9 kDa protein in BufCoV HKU26, a result of premature stop codon, and the 4.8 kDa protein was replaced with 3.2 and 5.1 kDa proteins in strains B1-24F and B1-28F, respectively, a result of frameshift deletion mutation (Lau et al., Reference Lau, Tsang, Ahmed, Alam, Ahmed, Wong, Yuen and Woo2016).
Sheep (Ovis aries)
Bovine-like CoVs have been identified as a cause of enteritis and neonatal mortality in domestic sheep in several countries worldwide (Tzipori et al., Reference Tzipori, Smith, Makin and McCaughan1978; Harp et al., Reference Harp, Myers, Rich and Gates1981; Pass et al., Reference Pass, Penhale, Wilcox and Batey1982; Reinhardt et al., Reference Reinhardt, Zamora, Tadich, Polette, Aguilar, Riedemann and Palisson1995). However, the prevalence of infection was mostly low and of minor importance. Among 545 diarrhoeic lambs originated from 12 sheep flocks in southern Idaho and western Montana, USA, bovine-like CoVs were detected in lambs of only one flock (Harp et al., Reference Harp, Myers, Rich and Gates1981). As well, CoVs were detected in six (out of 98; 6.1%) dead lambs but not in healthy lambs and ewes in Chile (Reinhardt et al., Reference Reinhardt, Zamora, Tadich, Polette, Aguilar, Riedemann and Palisson1995). Two reports from Spain and Turkey demonstrated that Cryptosporidium parvum, Giardia intestinalis, Escherichia coli and rotavirus – but not CoV – were the leading causes of neonatal diarrhoea and mortalities in domestic sheep (Muñoz et al., Reference Muñoz, Alvarez, Lanza and Cármenes1996; Ozmen et al., Reference Ozmen, Yukari, Haligur and Sahinduran2006). A nationwide screening of BCoV-specific antibodies in Swedish sheep has demonstrated that 19% of all sera samples were positive. However, it was expected that these antibodies originated from contact with infected cattle faeces rather than a natural infection with bovine-like CoV (Tråvén et al., Reference Tråvén, Carlsson, Lundén and Larsson1999).
Goat (Capra aegagrus hircus)
Few studies have investigated the role of bovine-like CoV in goat kids with neonatal diarrhoea. Examination of faecal samples from diarrhoeic and non-diarrhoeic kids (1–45 days old) in Spain has demonstrated complete absence of CoVs (Muñoz et al., Reference Muñoz, Alvarez, Lanza and Cármenes1996). While CoVs were detected in the faeces of one (out of 19; 3.3%) kid with neonatal enteritis in Turkey by immunohistochemistry (IHC). The positive reaction was obvious at the crypt epithelium of small intestine and in the submucosal macrophages (Ozmen et al., Reference Ozmen, Yukari, Haligur and Sahinduran2006). A large-scale serosurveillance for viral diseases has been conducted in 144 native goat farms in South Korea between 2005 and 2006. Among 804 sera samples collected from five provinces of South Korea, CoV HI antibodies were only detected in eight samples (1%), as the least prevalent viral infection in two provinces, while it was entirely absent in the other districts (Yang et al., Reference Yang, Hwang, Kim, Kweon, Lee, Kang, Lee and Cho2008).
Camelids
Nowadays, six species of camelids exist. They are grouped into two tribes: Camelini that includes old world camels, and Lamini that contains new world camels. Two species of old world camels include dromedary or one-humped camel (Camelus dromedarius) and Bactrian or two-humped camel (Camelus bactrianus). The majority of old world camels (90%) are dromedaries, which inhabit the African horn, North Africa, Middle East and Southwestern Asia. The Bactrian camel has an estimated worldwide population of 1.4 million animals living in central Asia (mostly in China and Mongolia) (Frankin, Reference Frankin, Wilson and Mittermeier2011). Few wild dromedary and Bactrian camels are still living in Australia and Gobi Desert, respectively (Saalfeld and Edwards, Reference Saalfeld and Edwards2008; Animal Information Organization, 2018).
On the other hand, the new world camels include two wild species: guanaco (Lama guanicoe) and viçuna (Vicugna vicugna), and two domesticated species: llama (Lama glama) and alpaca (Vicugna pacos). All of these species are living in South America (mostly in the Andean highlands) and are used for food, textile and transport (Hirst, Reference Hirst2017). About 2.5–3.0 million llamas are currently distributing in Bolivia (70%), Chile, Peru, and Argentina. Similarly, a world population of 3.88 million alpacas are mostly concentrated in Peru (77%), Bolivia (13%), Chile and Argentina. Breeding of llama and alpaca for commercial uses is expanding outside South America, particularly in the USA, Australia and many European countries (Crossley et al., Reference Crossley, Barr, Magdesian, Ing, Mora, Jensen, Loretti, McConnell and Mock2010; Thomas and Morgan, Reference Thomas and Morgan2013; La Vigna Alpacas, Reference La Vigna2018).
Old world camelids
The role of dromedary camels (DCs) as the most likely source of human infection with MERS-CoVs is gradually establishing with continuous appearance of new evidence (Alagaili et al., Reference Alagaili, Briese, Mishra, Kapoor, Sameroff, Burbelo, De Wit, Munster, Hensley, Zalmout, Kapoor, Epstein, Karesh, Daszak, Mohammed and Lipkin2014; Muhairi et al., Reference Muhairi, Hosani, Eltahir, Mulla, Yusof, Serhan, Hashem, Elsayed, Marzoug and Abdelazim2016; Sabir et al., Reference Sabir, Lam, Ahmed, Li, Shen, Abo-Aba, Qureshi, Abu-Zeid, Zhang, Khiyami, Alharbi, Hajrah, Sabir, Mutwakil, Kabli, Alsulaimany, Obaid, Zhou, Smith, Holmes, Zhu and Guan2016; Miguel et al., Reference Miguel, Chevalier, Ayelet, Ben Bencheikh, Boussini, Chu, El Berbri, Fassi-Fihri, Faye, Fekadu, Grosbois, Ng, Perera, So, Traore, Roger and Peiris2017). Because MERS-CoV has never been identified in cattle (Hemida et al., Reference Hemida, Perera, Wang, Alhammadi, Siu, Li, Poon, Saif, Alnaeem and Peiris2013; Reusken et al., Reference Reusken, Ababneh, Raj, Meyer, Eljarah, Abutarbush, Godeke, Bestebroer, Zutt, Muller, Bosch, Rottier, Osterhaus, Drosten, Haagmans and Koopmans2013a, Reference Reusken, Haagmans, Müller, Gutierrez, Godeke, Meyer, Muth, Raj, Smits-De Vries, Corman, Drexler, Smits, El Tahir, De Sousa, van Beek, Nowotny, van Maanen, Hidalgo-Hermoso, Bosch, Rottier, Osterhaus, Gortázar-Schmidt, Drosten and Koopmans2013b) and has many unique antigenic and genetic characteristics that make it distinct from BCoVs (Hemida et al., Reference Hemida, Perera, Wang, Alhammadi, Siu, Li, Poon, Saif, Alnaeem and Peiris2013), it will not be discussed further in this review.
Although DCs are normal inhabitants of Northern and Eastern Africa and Southwestern Asia, the first report of bovine-like CoV infections in DCs came from the USA (Wünschmann et al., Reference Wünschmann, Frank, Pomeroy and Kapil2002). A case of severe lethal gastroenteritis was recorded in a 6-week-old female camel calf in Wisconsin. CoV particles with typical club-shaped projections were identified in the faeces of the diarrhoeic camel calf by EM. The virus showed specific reactivity with two MAbs, prepared against BCoV S and nucleocapsid (N) proteins, in the epithelial cells of colonic crypts by IHC. The nature and origin of the CoV were not elucidated; however, the role of interspecies transmission was proposed because individuals of other animals like horses, zebras and reindeer were kept with the camel calf in the same barn at the time of infection.
In 2013, a novel CoV, designated as DcCoV UAE-HKU23, was identified in 14 (4.8%) leftover DC faecal samples (four with diarrhoea) in the Central Veterinary Research Laboratory, Dubai, UAE. Complete genome analysis of DcCoV UAE-HKU23 in three faecal samples has classified the virus as a member of lineage A1 of Betacoronaviruses (Woo et al., Reference Woo, Lau, Wernery, Wong, Tsang, Johnson, Yip, Lau, Sivakumar, Cai, Fan, Chan, Mareena and Yuen2014). However, genetic distance analysis and codon usage bias of polymerase, S and N genes of DcCoV UAE-HKU23 strains have demonstrated that it is distantly separated from other members of Betacoronavirus 1 and may form a unique cluster within the group (Woo et al., Reference Woo, Lau, Fan, Lau, Wong, Joseph, Tsang, Wernery, Yip, Tsang, Wernery and Yuen2016; Fig. 1). Recombinant N protein of DcCoV UAE-HKU23 has been generated and utilized for the detection of the virus-specific antibodies in sera of 59 camels originated from Sudan, Saudi Arabia, Oman and Pakistan (Woo et al., Reference Woo, Lau, Wernery, Wong, Tsang, Johnson, Yip, Lau, Sivakumar, Cai, Fan, Chan, Mareena and Yuen2014). Antibodies to the recombinant protein were identified in 98.3 and 100% of the tested sera samples using IF and VN assays. Immunization of mice with the recombinant N proteins of DcCoV UAE-HKU23 and MERS-CoV has confirmed minimal cross-antigenicity between both viruses in Western blot (WB) and VN assays (Woo et al., Reference Woo, Lau, Fan, Lau, Wong, Joseph, Tsang, Wernery, Yip, Tsang, Wernery and Yuen2016). These results demonstrate that a Betacoronavirus, other than MERS-CoV, is circulating among DCs in different countries of Middle East and may contribute to pathological conditions of diarrhoea, particularly in camel calves.
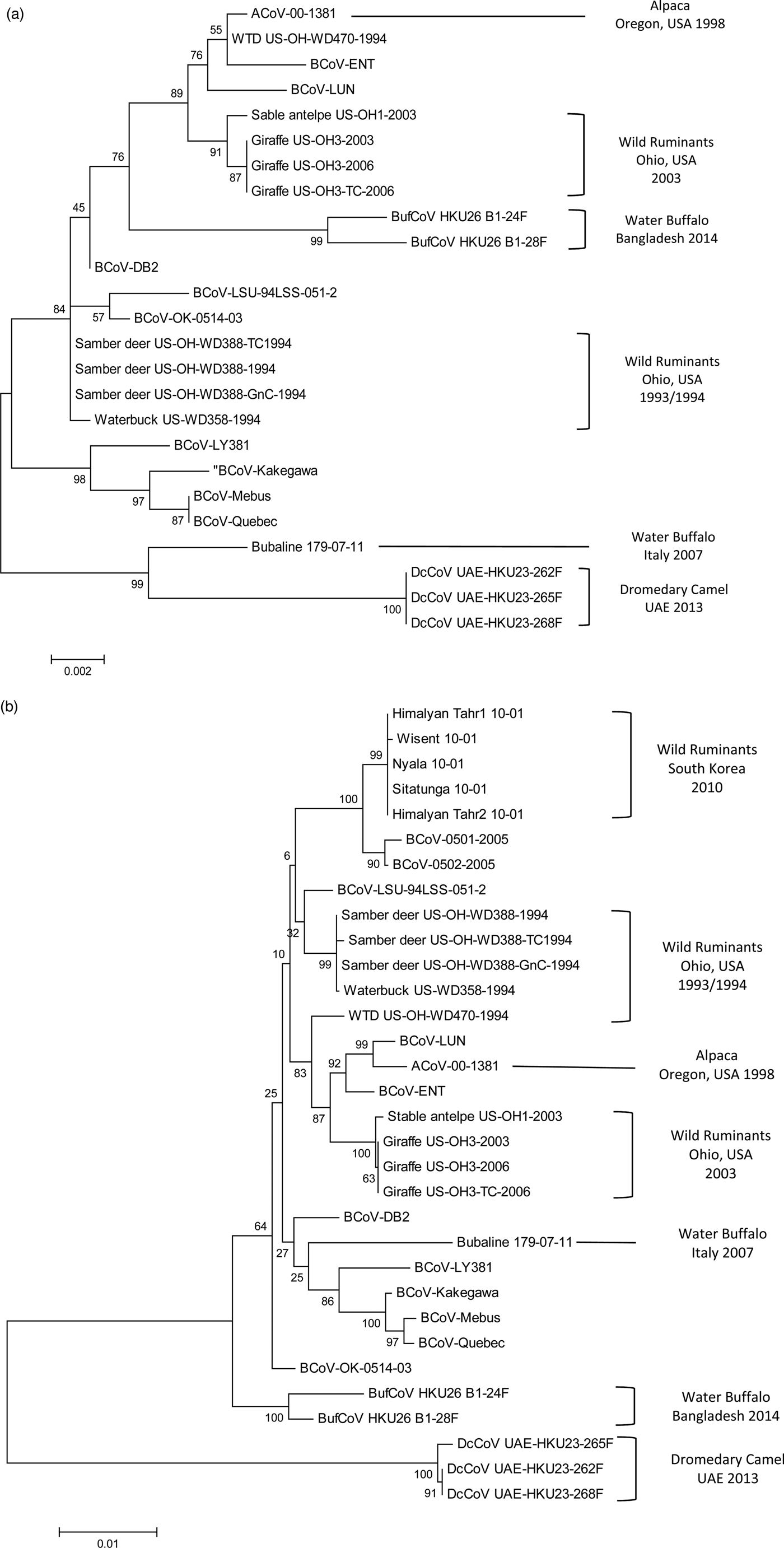
Fig. 1. Phylogenetic analysis of bovine-like coronaviruses based on the sequence of (a) nucleocapsid gene and (b) spike gene. Reference bovine coronaviruses were included for comparison purposes. Sequences were downloaded from GenBank (Table 3) and aligned together using Clustal W algorithm of MegAlign program, Lasergene software, version 3.18 (DNAStar, Madison, WI). Phylograms were constructed by MEGA 7.0 software using the maximum likelihood method. The strength of the tree was evaluated by bootstrapping of 1000 replicates. Bootstrap values are shown at the branch nodes of the tree. Bovine-like coronaviruses are grouped according to the type of animal(s), state or country of origin, and year of identification as indicated in the brackets at the right side. The scale bar at the bottom indicates the number of nucleotide changes per site.
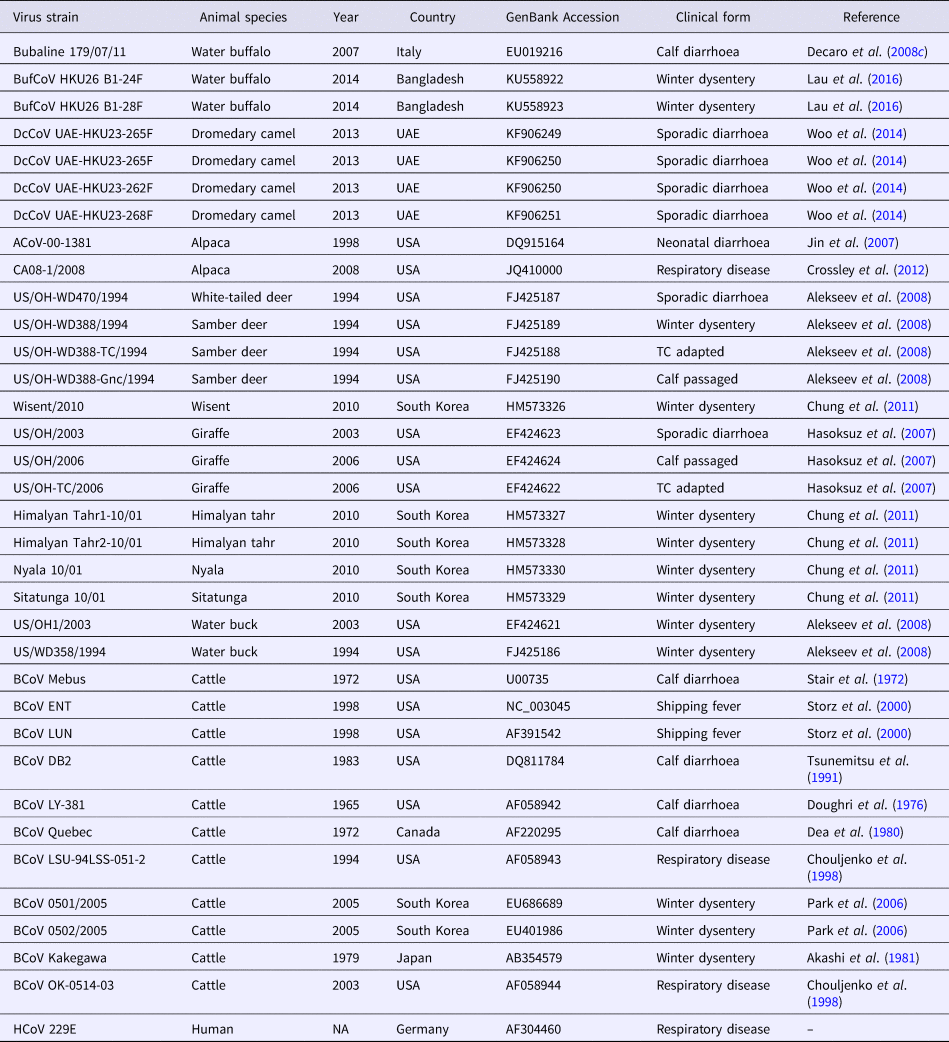
Table 3. Betacoronavirus 1 strains used in the phylogenetic analysis
Recently, three species of CoVs were identified in DCs in Saudi Arabia during a surveillance study that included 1309 nasal and rectal swab samples collected between May 2014 and April 2015 (Sabir et al., Reference Sabir, Lam, Ahmed, Li, Shen, Abo-Aba, Qureshi, Abu-Zeid, Zhang, Khiyami, Alharbi, Hajrah, Sabir, Mutwakil, Kabli, Alsulaimany, Obaid, Zhou, Smith, Holmes, Zhu and Guan2016). These CoVs included two Betacoronaviruses: MERS-CoV (group C) and camel HKU23-CoVs (group A), and one Alphacoronavirus (similar to human CoV 229E). All CoV-positive samples except three were nasal swabs, which indicates that the respiratory tract is the major shedding portal for CoVs in DCs. Regardless of MERS-CoV that has been detected in DCs in many countries worldwide, the HKU-23-CoV was detected in DCs in Dubai short time earlier (Woo et al., Reference Woo, Lau, Wernery, Wong, Tsang, Johnson, Yip, Lau, Sivakumar, Cai, Fan, Chan, Mareena and Yuen2014) and a closely related camelid Alphacoronavirus was isolated from alpacas in the USA in 2007 (Crossley et al., Reference Crossley, Barr, Magdesian, Ing, Mora, Jensen, Loretti, McConnell and Mock2010; Crossley et al., Reference Crossley, Mock, Callison and Hietala2012). The high prevalence of CoVs in the collected samples, the frequent co-infections and the lack of symptoms in most CoV-positive cases propose that these CoVs are enzootic in DCs, at least in Saudi Arabia, and that they may play an important role in CoV ecology.
New world camelids
CoV infection of llamas and alpacas was first recognized during an outbreak of severe diarrhoea in Oregon, USA in 1998. Among 39 pathogens detected, CoV was identified as the primary cause of the diarrhoeal complex in juvenile animals (crias) with a prevalence rate of 42%. Sick crias developed clinical symptoms similar to BCoV-infected cattle calves with variable degrees of severity and frequent deaths (Cebra et al., Reference Cebra, Mattson, Baker, Sonn and Dearing2003; Cebra, Reference Cebra2007). A single alpaca CoV (designated ACoV-00-1381) was isolated from the diarrhoeic samples by propagation in HRT-18 G cells. Sequence and phylogenetic analysis of the complete ACoV-00-1381 genome revealed close homology (>99.5%) with two BCoV strains involved in shipping fever pneumonia and enteritis in feedlot calves: LUN and ENT, respectively (Storz et al., Reference Storz, Stine, Liem and Anderson1996; Chouljenko et al., Reference Chouljenko, Lin, Storz, Kousoulas and Gorbalenya2001; Fig. 1). A common ancestral origin of alpaca CoV and both strains of BCoV was proposed (Jin et al., Reference Jin, Cebra, Baker, Mattson, Cohen, Alvarado and Rohrmann2007).
Bovine-like CoV was also acknowledged as a potential cause of neonatal diarrhoea in alpacas in two separate studies conducted in Cusco in southern Peruvian highlands. In the first, CoVs have been detected in three out of 14 (23%) of the alpaca crias using immunochromatography (López et al., Reference López, Chamorro and Garmendia2011). While in the second, the virus was detected in 20 out of 50 (40%) of the crias during an episode of diarrhoea in January and February 2010 using RT-PCR (Rojas et al., Reference Rojas, Manchego, Rocha, Fornells, Silva, Mendes, Dias, Sandoval, Pezo and Santos2016). All the cases in the second study showed co-infection with other (up to three) enteric pathogens including Eimeria spp., Cryptosporidium spp., E. coli and rotavirus. However, the exact role of CoV in disease development and progression was unclear.
CoV-associated diarrhoea in adult alpacas is generally a rare event and is supported mostly by anecdotal evidence. A single report has documented the identification of a CoV in faecal specimen collected from 4-year-old hembra (female alpaca) in Stillwater, Oklahoma (Genova et al., Reference Genova, Streeter, Simpson and Kapil2008). The virus was classified as a member of antigenic group 2 CoVs (later termed as Betacoronaviruses) as a result of its cross-reactivity with MAb specific for N protein of antigenic group 2 CoVs in IHC.
In contrast to all previous reports that correlate CoVs in alpaca with gastroenteritis, a novel CoV was retrieved from the lung tissue of a clinical case with alpaca respiratory syndrome (ARS) in California on October 2007. The CoV was isolated with the CRFK cell line and its identity was confirmed by transmission EM, sequence analysis of a short conserved fragment within the polymerase gene (Crossley et al., Reference Crossley, Barr, Magdesian, Ing, Mora, Jensen, Loretti, McConnell and Mock2010), and later by full-genome sequencing (Crossley et al., Reference Crossley, Mock, Callison and Hietala2012). The widespread nature of respiratory CoV in alpacas was verified through testing 40 sera samples taken from animals with a history of ARS and 167 sera samples randomly selected from alpacas with unknown disease history. The majority of samples with ARS history (60%) has shown antibody titres ≥1:16 in VN assay using the plaque-purified respiratory alpaca CoV. However, only 3% of the samples collected from animals with unknown history showed similar titres (Crossley et al., Reference Crossley, Barr, Magdesian, Ing, Mora, Jensen, Loretti, McConnell and Mock2010).
No consequent outbreaks of the respiratory alpaca CoV was recorded in the USA or elsewhere, which raised questions about the origin and circulation of the virus in the alpaca population. Genome sequencing of the respiratory alpaca CoV revealed close homology with the Alphacoronaviruses (particularly with HCoV-229E; 92.2%), unlike the enteric alpaca CoVs that were routinely identified as Betacoronaviruses. Comparison of S gene sequences from the respiratory alpaca CoV and a range of HCoV-229 isolates recovered from 1962 to 2003 showed close genetic homology with HCoV-229 strains isolated from the 1960s until the early 1980s. It was concluded that a common ancestor of HCoV-229E and respiratory alpaca CoV may have existed either in people or alpacas before 1960, and crossed the interspecies barrier to establish infection in the other species thereafter (Crossley et al., Reference Crossley, Mock, Callison and Hietala2012). The virus may have circulated among alpacas at low levels for decades before suddenly appearing during the 2007 outbreak. Explanation of the absence of further disease outbreaks in the subsequent years needs further investigation.
Bovine-like CoVs in captive and free-range wild ruminants
Deer (Cervidae)
Deer are one of the most widely distributed ruminant animals on the Earth. They constitute a natural component of the biological communities in all continents except Australia and Antarctica. All deer species are included in the family Cervidae with two subclasses: (1) old world deer (subfamily Cervinae), which contains 33 deer species including elk (wapiti), muntjac, chital, sika deer and samber deer, (2) new world deer (subfamily Capreolinae), which comprises 21 deer species including reindeer (caribou), pudu and European elk (moose) (Bertin, Reference Bertin2017). Breeding of deer (particularity in game farms) is currently a growing interest for venison production, which is characterized by high-protein content and low-cholesterol level (Smits, Reference Smits1991). To date, bovine-like CoVs were demonstrated in only six deer species, which are caribou/reindeer (Rangifer tarandus caribou) (Elazhary et al., Reference Elazhary, Frechette, Silim and Roy1981), elk/wapiti (Cervus elephus) (Smits, Reference Smits and Brown1992; Majhdi et al., Reference Majhdi, Minocha and Kapil1997; Daginakatte et al., Reference Daginakatte, Chard-Bergstrom, Andrews and Kapil1999), samber deer (Cervus unicolor), white-tailed deer (Odocoileus virginianus) (Tsunemitsu et al., Reference Tsunemitsu, El-Kanawati, Smith, Reed and Saif1995; Alekseev et al., Reference Alekseev, Vlasova, Jung, Hasoksuz, Zhang, Halpin, Wang, Ghedin, Spiro and Saif2008), sika deer (Cervus nippon yesoensis) (Yokoi et al., Reference Yokoi, Okazaki, Inahara and Hatama2009) and water deer (Kim et al., Reference Kim, Jang, Yoon, Noh, Ahn, Kim, Jeong and Kim2018).
Cariboo/reindeer
The evidence of bovine-like CoVs in Cariboo was demonstrated through a surveillance study that investigated the prevalence of antibodies to five bovine respiratory and enteric viruses in two caribou herds in Northern Quebec, Canada. While bovine viral diarrhoea virus was the most prevalent in both years, bovine-like CoV was the least prevalent (bovine parainfluenza 3 virus was absent from all samples). Among 30 samples collected in 1978 and 28 samples in 1979, CoV-specific antibodies were detected in four (13.3%) and zero (0%) samples in both years, respectively. The source of bovine-like CoV infection was unclear because both caribou herds had no direct contact with domestic ruminants for at least 25 years (Elazhary et al., Reference Elazhary, Frechette, Silim and Roy1981).
Elk/wapiti
CoVs have been frequently linked to enteritis and scouring in neonatal elk. The virus was isolated as the only pathogen in five out of 11 calves with diarrhoea (45.5%) in western North America (Smits, Reference Smits1991; Smits, Reference Smits and Brown1992). In a subsequent study, two CoV isolates were retrieved from faecal samples collected from 10-month-old captive elk calves with diarrhoea in the USA (Majhdi et al., Reference Majhdi, Minocha and Kapil1997). The virus isolates in the latter study (designated WY-28 and WY-29) showed a high degree of antigenic and genetic relation to BCoV, based on several observations: (1) comparable protein profiles as determined by SDS-PAGE, (2) similar transcriptional patterns in Northern blot analysis, (3) significant sequence homology (99%) between N gene sequence of both viruses, (4) cross-reactivity with MAb specific for S protein of BCoV in WB, and (5) ability of four MAbs specific for N protein of elk CoV, to detect BCoV in intestinal tissues by IHC (Majhdi et al., Reference Majhdi, Minocha and Kapil1997; Daginakatte et al., Reference Daginakatte, Chard-Bergstrom, Andrews and Kapil1999).
Samber and white-tailed deer
Infection of samber deer with bovine-like CoVs was only recorded during an outbreak of diarrhoea that occurred in a wild animal park in southern Ohio, USA in the winter season of 1993/1994. Affected animals showed severe symptoms of bloody diarrhoea, which resemble WD in adult cattle, with a mortality rate of 30%. In another wild animal park in north central Ohio, episodes of watery diarrhoea in white-tailed deer were also monitored throughout 1994. CoV particles were identified in faecal samples collected from three samber deer and one white-tailed deer using immunocytochemistry and antigen detection ELISA. Two CoV isolates (KI-D2 from samber deer and WTD from white-tailed deer) were recovered in the Cariboo HRT-18 cell line. Both isolates demonstrated close relationship with several BCoV strains causing CD and WD based on similar biologic (haemagglutination and receptor-destroying activities) and antigenic reactivity in HI, VN, IF and WB tests (Tsunemitsu et al., Reference Tsunemitsu, El-Kanawati, Smith, Reed and Saif1995). Analysis of the full genome sequence of both isolates (either cell culture-adapted or gnotobiotic calf-passaged) confirmed the marked similarity with BCoVs, particularly LUN and ENT strains isolated in 1998 (Fig. 1b), with no specific genetic markers that can discriminate KI-D2 and WTD from BCoV strains (Alekseev et al., Reference Alekseev, Vlasova, Jung, Hasoksuz, Zhang, Halpin, Wang, Ghedin, Spiro and Saif2008). Additionally, 30 sera samples were acquired from free-range healthy white-tailed deer located in a third wild animal park in Ohio. Two samples (6.6%) were seropositive against both isolates and several BCoV strains in IF assay (Tsunemitsu et al., Reference Tsunemitsu, El-Kanawati, Smith, Reed and Saif1995).
Sika deer
This type of deer is a native species in Hokkaido, Japan. A single observation of potential infection with bovine-like CoV was reported in a serosurveillance study that evaluated the prevalence of antibodies to eight bovine viruses in sika deer. Serum samples were collected from a farm of sika deer in eastern Hokkaido between June 2006 and September 2007 and from wild animals during the hunting season of 2000. Among 179 farmed deer, only two (1.1%) samples were reactive to BCoV in VN assay. Unfortunately, not all samples collected from wild sika deer (no = 97) were tested for BCoV (Yokoi et al., Reference Yokoi, Okazaki, Inahara and Hatama2009).
Water deer
The evidence of water deer (Hydropotes inermis) infection with bovine-like CoVs was not demonstrated in captive animals during a study that extended between 2010 and 2012 in South Korea (Kim et al., Reference Kim, Choi, Yoon, Woo, Chung, Kim and Shin2014). However, in a recent study that involved 77 nasal swab samples, collected during 2016 and 2017 from non-captive water deer living in Chungnam Wild Animal Rescue Center, South Korea, three positive samples (3.9%) were obtained. The phylogenetic analysis, based on a partial sequence of the polymerase gene, indicated close relationship of the three water deer CoVs with BCoV (99.2%). Complete genome sequencing of a single water deer CoV strain (designated W17-18) revealed close relationship to BCoV and other bovine-like CoVs (homology >98%) with minor unique genetic characteristics of the non-structural protein 4.8 kDa. This report is the only one that proposes the implication of bovine-like CoVs in respiratory diseases of wild ruminants (Kim et al., Reference Kim, Jang, Yoon, Noh, Ahn, Kim, Jeong and Kim2018).
Wild cattle
Similar to domestic cattle, wild bovidae have also been associated with CoV-induced scours in calves and WD in adults. During the winter season of 1979–1980, an outbreak of diarrhoea occurred among a group of musk oxen (Ovibox moschatus) in Whipsnade zoological park in Bedfordshire, UK. CoVs were identified in the faecal samples collected from diseased animals using EM. Unfortunately, virus strains were not isolated or characterized due to technical limitations (Chasey et al., Reference Chasey, Reynolds, Bridger, Debney and Scott1984). CoVs are also among the common pathogens isolated from bison (Bison bison) calves with diarrhoea (Haigh et al., Reference Haigh, Mackintosh and Griffin2002). However, the actual prevalence of CoVs in bison is underestimated because bison mask the clinical signs of the disease syndrome (Berezowski, Reference Berezowski2001). CoVs were also demonstrated in the faeces of adult wisent – European bison (Bison bonasus) suffering from severe diarrhoea in the National Zoo of South Korea. The outbreak started in a wisent, albeit no cattle were housed in the zoo and no cattle farms existed in the vicinity, and spread to other wild ruminants in the park. All identified CoVs were quite similar in terms of nucleotide and deduced amino acid sequence. They also showed high level of genetic relationship (99.4–99.5%) to BCoVs isolated in South Korea after 2004, particularly BCoV-0501 and BCoV-0502 strains (Fig. 1b). It was concluded that all bovine-like CoVs characterized in this study were replicates of a single strain that has no restricted host specificity. This strain may originate, via an unclear pathway, from the same ancestor of BCoV-0501 and BCoV-0502 strains of cattle (Chung et al., Reference Chung, Kim, Bae, Lee and Oem2011). More recently, a serological study was conducted in a population of wood bison (B. bison athabascae) that were reintroduced in the wild in southwestern Youkon, Canada since 1988. Antibodies to BCoV were demonstrated in only two out of 31 (7%) of the animals tested (Harms et al., Reference Harms, Jung, Andrew, Surujballi, VanderKop, Savic and Powell2019).
Antelopes
The antelope is a deer-like mammal that belongs to the family Bovidae but is not cattle, buffalo, sheep or goat. Antelope is not a taxonomic name rather than a common term that collects a variety of grazing ruminants characterized by long legs and permanent horns. These old world animals are indigenous to Africa and Euroasia and may be found in parts of the Americas (Estes, Reference Estes2017). To date, the role of bovine-like CoVs in CD and WD was only described in four species of antelopes including waterbuck (Kobus ellipsiprymnus) (Chasey et al., Reference Chasey, Reynolds, Bridger, Debney and Scott1984; Tsunemitsu et al., Reference Tsunemitsu, El-Kanawati, Smith, Reed and Saif1995), sitatunga (Tragelaphus spekei) (Chasey et al., Reference Chasey, Reynolds, Bridger, Debney and Scott1984; Chung et al., Reference Chung, Kim, Bae, Lee and Oem2011), nyala (Tragelaphus angasii) (Chung et al., Reference Chung, Kim, Bae, Lee and Oem2011) and sable antelope (Hippotragus niger) (Hasoksuz et al., Reference Hasoksuz, Alekseev, Vlasova, Zhang, Spiro, Halpin, Wang, Ghedin and Saif2007).
Waterbuck
In the beginning of 1982, an outbreak of watery diarrhoea was suddenly erupted in a herd of nine waterbucks in the Cotswold wildlife park in Oxfordshire, England. Four animals (two adults and two calves) were lost and bovine-like CoVs were identified in the faeces of affected animals using EM and ELISA. Attempts to propagate these viruses in HRT-18 cell culture were unsuccessful. Additionally, experimental infection of gnotobiotic cattle calves with the faecal materials of dead waterbucks did not reveal clinical reproduction of the disease. The inoculated calves did not shed the virus in their faeces, did not develop antibody responses and were not immune against experimental challenge with BCoV field strain (Chasey et al., Reference Chasey, Reynolds, Bridger, Debney and Scott1984). Infection of waterbuck with bovine-like CoVs was further confirmed in another epizootic outbreak of diarrhoea that occurred in Ohio wild animal park in 1993 and 1994. The aetiologic virus (designated KI-WB) has been isolated in cell culture and has close biologic and antigenic relation to BCoVs (Tsunemitsu et al., Reference Tsunemitsu, El-Kanawati, Smith, Reed and Saif1995). Unlike the virus detected in the UK in 1982, the gnotobiotic and colostrum-deprived calves experimentally infected with KI-WB strain developed signs of acute diarrhoea and excreted the virus in their faeces and nasal secretions.
Sitatunga
CoVs were identified in faecal samples collected from sitatunga on two different occasions. The first was during an outbreak of diarrhoea in Whipsnade Zoological Park in the winter season of 1979/1980 (and again in February 1984) using EM and ELISA (Chasey et al., Reference Chasey, Reynolds, Bridger, Debney and Scott1984). The second occurred during an outbreak that affected four animal species, including sitatunga, in the National Zoo of South Korea in 2010. Sitatunga was the only animal that showed both enteric and respiratory symptoms. Among 16 affected animals in the park, rectal swabs were taken from three adult sitatunga (older than 2 years) and all of these samples showed the characteristic CoV particles by EM (Chung et al., Reference Chung, Kim, Bae, Lee and Oem2011).
Nayla
Two nayla animals were affected during the diarrhoea epidemic that occurred in the National Zoo of South Korea in 2010. The CoV particles detected in the faeces of affected animals were quite similar to those identified in wisent, sitatunga and Himalayan tahr, suggesting all of these are host-range variants of the same strain (Chung et al., Reference Chung, Kim, Bae, Lee and Oem2011).
Sable antelope
An outbreak of diarrhoea occurred in several ruminant species in an Ohio wild animal park in 2003. The outbreak started in a sable antelope and spread 1–2 weeks later to giraffes that were housed in a separate barn 0.5 miles away. A single faecal sample was obtained from the sable antelope and was tested by immunocytochemistry using BCoV-specific antisera. CoV particles were detected in the faecal sample, but, unlike the giraffe isolates, the virus strain could not be adapted to cell culture or to induce productive infection in gnotobiotic calves (Hasoksuz et al., Reference Hasoksuz, Alekseev, Vlasova, Zhang, Spiro, Halpin, Wang, Ghedin and Saif2007). Genetic analysis revealed close relatedness with the giraffe isolates, several BCoVs, and to a lesser extent with KI-D2, WTD and KI-WB strains isolated from samber deer, white-tailed deer and waterbuck, respectively (Alekseev et al., Reference Alekseev, Vlasova, Jung, Hasoksuz, Zhang, Halpin, Wang, Ghedin, Spiro and Saif2008; Fig. 1).
Giraffe (Giraffa camelopardalis)
Giraffes are the tallest animals and the largest ruminants on the Earth. They mostly inhabit Africa and are considered vulnerable to extinction, with an overall worldwide population of 97,000 animals (Milman, Reference Milman2017). A single report described isolation and characterization of bovine-like CoVs from giraffes during the outbreak of diarrhoea in an Ohio wild animal park in 2003 (Hasoksuz et al., Reference Hasoksuz, Alekseev, Vlasova, Zhang, Spiro, Halpin, Wang, Ghedin and Saif2007). Bovine-like CoVs were identified in the faeces of three (two males and one female) giraffes by immunocytochemistry. The virus was successfully adapted and isolated from the faecal extract of a 24-year-old male giraffe with HRT-18 cell culture. The virus isolate (designated as GiCoV-OH3) revealed close biologic, antigenic and genetic relationship with different strains of BCoV, principally with the enteric strains ENT and DB2.
Although infection in giraffes was conclusive from the outbreak progression in the wild animal park, the definite source of this outbreak remains uncertain. The infection may have originated from wild animals in the park, which harbour the virus without clinical signs, or from a nearby cattle farm. The latter possibility was supported by the ability of GiCoV-OH3 strain to induce severe diarrhoea when orally administrated into a gnotobiotic cattle calf, with virus shedding that lasts for 2–3 days. The risk of virus transmission between cattle and giraffes seems important, particularly in Africa where grazing regions of both animals overlap. This may allow virus evolution and potential spread of new CoV strains to remote regions upon change in pastures, game farming and transfer of wild animals to zoological parks.
Himalayan tahr (Hemitragus jemlahicus)
Tahr is a wild species of goat native to the Himalayan mountains in Central Asia. They are specially adapted to life on the mountain slopes and are now considered a near-threatened species (Bhatnagar and Lovari, Reference Bhatnagar and Lovari2008). During the epizootic outbreak of WD that affected several ruminant species in the National Zoo of South Korea in 2010, three Himalayan tahrs presented signs of weakness, depression, anorexia, bloody diarrhoea and dehydration. Bovine-like CoVs were demonstrated in the faeces of affected animals using EM and their genetic characteristics were analysed (Chung et al., Reference Chung, Kim, Bae, Lee and Oem2011).
Conclusion
Bovine-like CoVs are widely distributed in domestic ruminants other than cattle and in captive and free-range wild ruminant species. The majority of bovine-like CoVs have confirmed close relationship with different BCoV strains that produce gastroenteritis in neonatal calves and lactating cows, and respiratory disease complex in growing and steer calves. It is generally accepted today that bovine-like CoVs are host-range variants of BCoV, which is crossing the interspecies barriers on a regular basis. Transmission of BCoV variants from cattle to other ruminants and vice versa allows persistence of the infection in nature, recurrent emergence of epidemics and continuous evolution of the virus. Wild ruminants, in particular, are not confined to discrete geographic regions and are always moving to seek new pastures, to escape from predators, and to find mates. Human activities, including – but not restricted to – deforestation, over-hunting and game farming, also force wild animals to change habitats. Continuous movement of wild animals promotes virus transmission to new terrestrial areas, and, importantly, provides a window for the virus to adapt to new hosts and to develop into novel strains.




