



Background
Leber hereditary optic neuropathy (LHON) is a relatively rare, but highly debilitating and bilaterally blinding disease.Reference Newman, Carelli, Taiel and Yu-Wai-Man1,Reference Carelli, Carbonelli and de Coo2 The maternally-inherited disorder leads to retinal ganglion cell dysfunctionReference Gueven3 and portends a poor visual prognosis.Reference Carelli, Carbonelli and de Coo2 LHON is generally a non-syndromic disease and most often affects otherwise healthy young people, with a male predominance of approximately 86%.Reference Wallace, Singh and Lott4 Patients typically present with relatively rapid progression of painless central vision loss, and the vast majority (97%) develop bilateral nerve involvement within one year of onset.Reference Carelli, Carbonelli and de Coo2 Classic findings on ocular examination include pale optic nerves, most prominent temporally due to a predilection of the disease for the papillomacular bundle. The reported incidence of LHON ranges between 1 in 30,000 and 1 in 50,000 people.Reference Carelli, Carbonelli and de Coo2
There are three classic mutations associated with LHON: m.3460G>A, m.11778G>A, and m.14484T>C. These mutations are found within the coding sequence of mitochondrially encoded genes of NADH-ubiquinone oxidoreductase core subunits 1, 4, and 6 (MT-ND1, -ND4, -ND6, respectively). The most common of these mutations is m.11778G>A, accounting for 75% of all LHON cases in North America.Reference Mackey, Oostra and Rosenberg5 As a mitochondrial mutation, male patients do not transmit the gene to their children, but all children of women with LHON will carry the mutation (with the exception of maternal gonadal mosaicism, in which not all children will inherit the mutation). Most LHON carriers and patients have homoplasmic levels of the mutation allele, meaning that all their mitochondrial DNA is affected by the mutation. However, because LHON mutations demonstrate incomplete penetrance, not all people with a mutation in these genes will develop LHON. Only about 50% of males and 10% of females harbouring known LHON mutations develop retinal ganglion cell loss.Reference Yu-Wai-Man, Vortuba, Moore and Chinnery6 Tissue mosaicism may also contribute to this finding, as peripheral blood levels of genetic material may not represent levels in all tissues.
Treatment options for LHON are very limited. Options such as “mitochondrial cocktails”, idebenone, and gene therapy have been investigated, but there remains no universally endorsed therapy to protect or improve vision.Reference Zuccarelli, Vella-Szijj, Serracino-Inglott and Borg7 A major hurdle to finding an appropriate and effective treatment for LHON remains the lack of data describing its natural history with respect to visual loss and potential for recovery. Although some studies are being conducted to fill this knowledge gap,Reference Lam, Feuer and Schiffman8 few studies have been conducted in Canada to determine the incidence and natural course of disease seen in this part of the world. The purpose of this study is to provide insight into potential LHON mutations found in Southwestern Ontario and to describe in detail the demographics and clinical characteristics of those patients examined at one large academic centre.
Methods
This study was conducted under institutional research ethics board approval and adhered to the tenets of the Declaration of Helsinki. It was designed to specifically look at genetic mitochondrial mutations on the MT-ND1, -ND4, and -ND6 genes causing LHON in Southwestern Ontario, Canada. The mutations were detected by the reference laboratory for the region at the London Health Sciences Centre (LHSC) Molecular Diagnostic Program. The study was conducted as a retrospective genetic and clinical chart review to provide an overview of LHON in Southwestern Ontario over a 5-year time period. All data were entered into a REDCap database and anonymized.
Patients included in the study were originally identified by obtaining a complete list of patients in whom any mitochondrial mutation was detected between January 2015 and January 2020 at LHSC. From this list, any patient who had a mutation detected on the MT-ND1, -ND4, or -ND6 genes was included. Although cases of LHON have been reported secondary to mutations in locations outside of the MT-ND genes (e.g., Mitochondrial cytochrome B and ATP6), these were not included in the study due to limited availability of comprehensive chart data, which could potentially lead to inclusion of patients who were unlikely to have LHON.
A clinical chart review was attempted on each patient identified through the genetics database. For those patients in which a chart was accessible, information abstracted included patient age, sex, identified mutation, presenting and stable visual acuity, time between symptom onset in each eye, presence of a relative afferent pupillary defect (RAPD), and optic nerve findings. Further information also included optical coherence tomography (OCT) and visual field findings at presentation and any abnormalities on neuroimaging. Patients’ family history, other health issues, and psychiatric history were also documented. For those patients in whom a chart could not be obtained, the only data that could be collected was age at the time of genetic testing, sex, implicated gene, mutation, amino acid substitution, percentage homoplasmy, and American College of Medical Genetics (ACMG) score. In this scoring system, 1 indicates a pathogenic mutation, 2 indicates likely pathogenic, and 3 indicates a variant of unknown significance.
As the laboratory in LHSC is the reference laboratory for Southwestern Ontario, the denominator for patients captured in this study was taken as the population of the South West Health Region from the 2016 census.9 Data from the larger subset of patients on whom limited information was available were pooled to provide statistical averages. Data obtained from patients who underwent chart review were analyzed primarily in a qualitative manner given the limited number of patients.
Results
Genetics Review
Of the 1517 patients assessed within the genetics database at LHSC who had been screened for mitochondrial genetic variants, 63 patients had a mutation in one of the LHON-associated MT-ND genes. Of those patients, 45 had mutations in one of the commonly sited loci (m.3460G>A, m.11778G>A, or m.14484T>C). The remaining 18 patients had other mutations on these genes.
Data obtained for the 63 patients were divided for analysis purposes. The three analysis groups included (1) patients with classic LHON mutations, (2) patients with possible novel LHON mutations, and (3) all patients combined. Approximately 50% of patients were male in all three groups. Overall, the majority (55.6%) of patients were between the ages of 10 and 39 at the time of genetic testing, although there was a significant proportion (20.6%) aged 60–69 in all three groups (Table 1). There were no patients with age greater than 70 at the time of testing. Among patients with classic LHON mutations, 6.7% had the m.3460 mutation, 44.4% the m.11778 mutation, and 48.9% the m.14484 mutation. Of the novel mutations, there were no two patients with the same mutation. Overall, 15.9% had a mutation on the MT-ND1 gene, 38.1% on the MT-ND4 gene, and 46.0% on the MT-ND6 gene.
Table 1: Characteristics of patients tested at LHSC with mutations found on MT-ND1, -ND4, and -ND6 genes

* Patients included in chart review.
** American College of Medical Genetics (ACMG) Score: 1 = pathogenic mutation, 2 = likely pathogenic, 3 = variant of unknown significance.
The population of the South West Ontario Health Region in 2016 was 953,261.9 Considering all possible mutations, the incidence of LHON was calculated to be 1 in 77,000 per year. Including only the classic mutations proven to cause LHON (m.3460, m.117787 and m.14484), the incidence was calculated to be 1 in 106,000 per year.
Chart Review
Of the 63 patients identified, only 14 patients had clinical charts accessible for review. Eleven of these patients had a classic LHON mutation and 3 had possible novel mutations on implicated LHON genes, in the context of either optic neuropathies or a family history of LHON. Twelve patients had symptomatic disease. All but one of the symptomatic patients had bilateral optic neuropathies, and 9 of them had classic LHON mutations (3 had possible novel mutations). Seven of the symptomatic patients were males, and 5 females. The two asymptomatic carriers were females. All but two of the symptomatic patients were under the age of 40 at the time of symptom onset. Nine patients had final visual acuity equal to or worse than 20/200 in at least one eye, and 6 of those had 20/400 or worse in both eyes (Table 2).
Table 2: Demographic characteristics and ophthalmological findings in patients tested at LHSC with LHON and/or potentially novel mutations associated with optic neuropathies

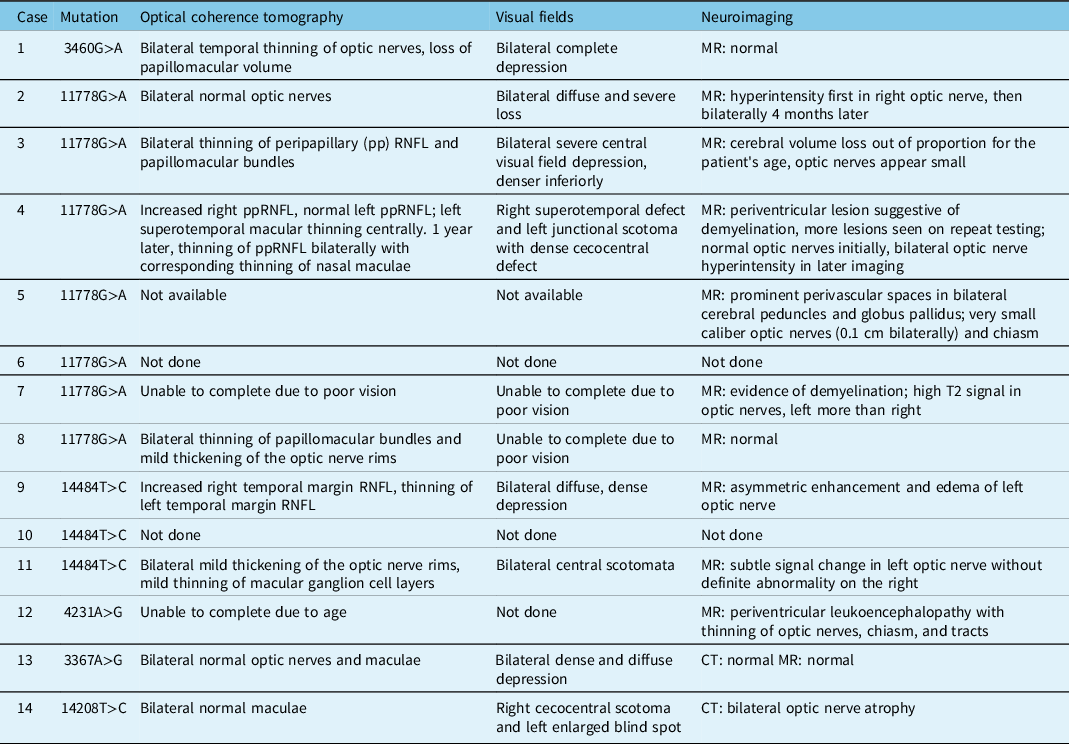
The majority of patients had pale optic nerves, although two patients with significantly reduced visual acuities had normal appearing optic nerves. These patients also had normal OCT imaging of the optic nerves, whereas all patients who had optic disc pallor showed thinning of their retinal nerve fibre layer (RNFL) when an OCT was performed. Two patients had hyperemic discs and thickening of the optic nerve rim on OCT at presentation. Of the patients who had visual fields, these showed significant, often diffuse, depression in the affected eyes. Some patients were unable to complete OCT or visual fields due to severe loss of vision. While some patients demonstrated normal neuroimaging, others were noted to have hyperintensity of the optic nerves on MRI or small caliber optic nerves. Those patients with a concurrent diagnosis of multiple sclerosis (MS) also had evidence of demyelination on their MRI (Table 3).
Table 3: Ancillary tests at presentation in patients tested at LHSC with LHON and/or potentially novel mutations associated with optic neuropathies
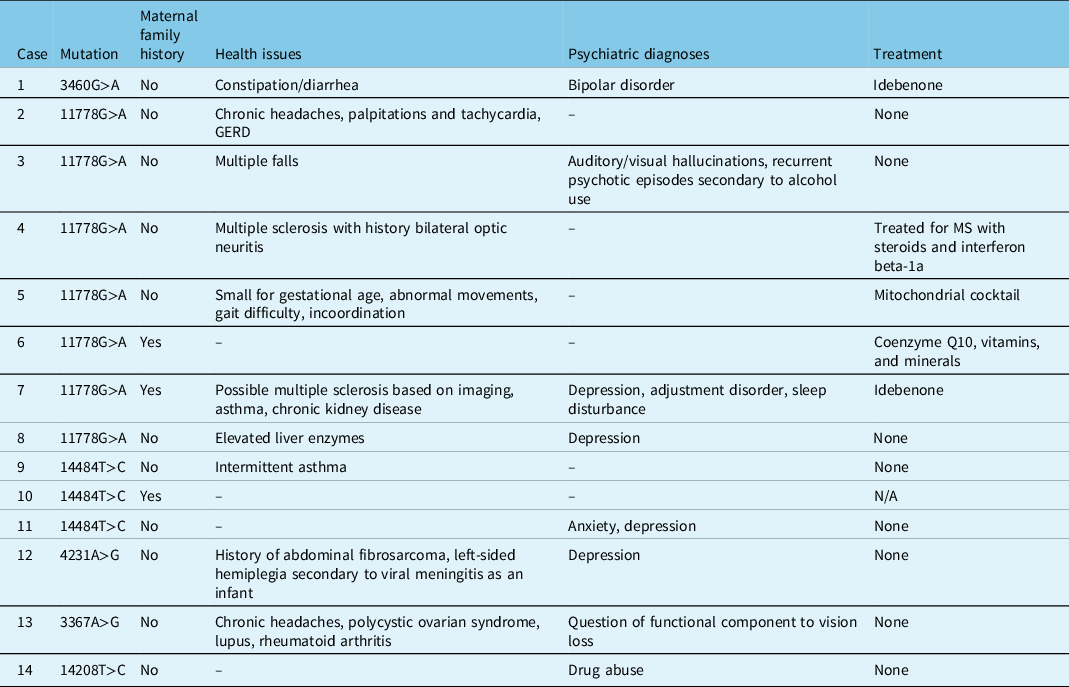
Three patients had a maternal family history of LHON, two of whom were asymptomatic and one (Patient 7) who had significant disease (Table 4). With a few exceptions, most patients were otherwise relatively healthy (Table 4). Patient 4 and 7 both had a concurrent diagnosis of MS, and Patient 5 had other neuromuscular symptoms of unknown etiology. Patient 12 suffered from hemiplegia secondary to a viral meningitis as a child. Psychiatric diagnoses were seen in 8 patients, with a suspected component of non-organic vision loss in Patient 13 (Table 4).
Table 4: Family history, medical history, and treatment in patients tested at LHSC with LHON and/or potentially novel mutations associated with optic neuropathies
Discussion
In the literature, the most common LHON mutation is at the m.11778 locus on MT-ND4.Reference Mackey, Oostra and Rosenberg5 However, in the genetics review presented here, it accounted for only 44.4% of classic mutations and 31.7% of all mutations. The most common mutation was at the m.14484 locus on MT-ND6, accounting for 48.9% of classic mutations and 34.9% of all mutations (Table 1). This may be accounted for by the known founder effect of French Canadian families with the m.14484 mutation.Reference Laberge, Jomphe and Houde10 In the small chart review, the m.11778 mutation accounted for the majority (50%) of mutations. This mutation is also generally associated with the worst visual outcome.Reference Newman, Carelli, Taiel and Yu-Wai-Man1 Four of the 7 patients in the chart review with this mutation ended up with vision of counting fingers (CF) or worse in at least one eye (Table 2). The mutation at the m.14484 locus has been shown to have the best visual prognosis, showing improvement in up to 50% of patients.Reference Yu-Wai-Man, Vortuba, Moore and Chinnery6 There were two symptomatic patients (Patients 9 and 11) in our series who had a mutation at this locus, and both demonstrated visual deterioration over time rather than improvement (Table 2).
Overall, female representation and older age at presentation were more common in this series than previously reported in the literature. The male-to-female ratio of LHON mutations in our genetics review was 1:1 and in the chart review of symptomatic patients was 1.4:1. This unexpected distribution may be due to the small sample size. However, there is growing evidence to suggest that the incidence of LHON in women is higher than previously thought. A large international study looking at the demographics of LHON determined a male-to-female ratio of 3:1,Reference Poincenot, Pearson and Karanjia11 lower than the previously reported 5:1 ratio. They also found that below the age of 5 or above the age of 45 at time of onset, the sex ratio was 1:1.Reference Poincenot, Pearson and Karanjia11 A significant proportion (40%) of the patients in our genetics review were 40 or older at the time of genetic testing, and this may explain the higher female ratio in our study. However, given that the genetics review could only provide age at time of testing, it is not a true representation of age at symptom onset. Our chart review found that only two of the 12 symptomatic patients had symptom onset after the age of 35. The 2020 large international study found that 10% of patients developed symptoms after age 50.Reference Poincenot, Pearson and Karanjia11
The vast majority of mutations implicated in LHON are found in mitochondrial DNA on the genes coding complex I subunits of the respiratory chain (NADH-ubiquinone oxidoreductase).Reference Catarino, von Livonius and Priglinger12 Mutations causing amino acid substitution lead to dysfunction in the respiratory complex I, whose function is to process oxygen and create energy within the cell. Disruption of this process translates to subacute loss of retinal ganglion cells and their axons, which comprise the optic nerve.Reference Kirches13 While many of the novel mutations proposed here (Table 1) are not previously documented to cause LHON, they do all cause an amino acid substitution within the respiratory complex I, and therefore have a plausible mechanism for causing disease. Given the lack of clinical information available on most of the 63 patients, it cannot be ascertained if they all had optic neuropathies or whether other symptomology or family history may have prompted genetic testing. There are multiple known mutations found on the MT-ND1, -ND4, and -ND6 genes that are associated with other conditions such as Leigh syndrome, MELAS, and Parkinson’s disease.Reference Manickam, Michael and Ramasamy14 For this reason, all possible novel mutations included in the genetics review were searched in the literature to determine any previously identified associations. Multiple novel mutations in our genetics review were found in the literature in cases of LHON, supporting that these mutations are likely pathogenic. These include the m.4024A>T,Reference Korkiamäki, Kervinen, Karjalainen, Majamaa, Uusimaa and Remes15 m.3866T>C,Reference Zhou, Qian and Zhang16,Reference Ji, Zhang and Lu17 and m.14459G>AReference Tarnopolsky, Baker, Myint, Maxner, Robitaille and Robinson18–Reference Jun, Brown and Wallace20 mutations (Table 1). The only mutation that yielded an association outside of LHON was m.11240C>T, which has been linked to Leigh syndrome in one previous case.Reference Xu, Li and Du21 All patients in the small chart review with novel mutations demonstrated optic neuropathies and homoplasmy of their mutation (Table 1), which is consistent with LHON.Reference Yu-Wai-Man, Vortuba, Moore and Chinnery6 These mutations were m.4231A>G, m.3367A>G, and m.14208T>C (Table 2). None of these have previously been documented to be of clinical significance in the literature.
The ophthalmological findings of patients in the chart review are consistent with what has previously been reported in the literature. Most patients demonstrated optic nerve pallor, primarily in the temporal quadrant. Two patients had hyperemic nerves, which can be seen in the early stages of disease.Reference Shemesh, Sood and Margolin22 Patients who underwent OCT of the optic nerves demonstrated corresponding thinning or thickening of the nerves, depending on the timing of testing. On MRI, patients showed a range of normal optic nerves, hyperintense signal, edema, and thinning; all features which have been previously reported in cases of LHON.Reference Blanc, Heran, Habas, Bejot, Sahel and Vignal-Clermont23 Two patients demonstrated normal optic nerves, which can be seen at presentation, but eventually optic atrophy is expected to develop in all patients with symptomatic LHON.
Although LHON is generally nonsyndromic, various systemic findings have been reported in patients with LHON. Two female patients (Patients 4 and 7) in this review suffered from a concurrent diagnosis of MS, compatible with numerous reports and case series in the literature describing this co-phenomenon. Harding (1992) described 11 patients with LHON and MS,Reference Harding, Sweeney and Miller24 hypothesizing that the presence of an LHON mutation in the mitochondria can be an instigator for MS. This is most often seen in female patients, and a previous study found that 45% of females with the m.11778 mutation have an MS-like illness,Reference Riordan-Eva, Sanders, Govan, Sweeney, Da Costa and Harding25 as demonstrated in Patients 4 and 7. Female patients with LHON who have no MS-like symptoms are still more likely to have MRI abnormalities suggestive of MS.Reference Harding, Sweeney and Miller24
Three other patients in the chart review suffered from serious medical illnesses. Patient 5 was small for gestational age with abnormal movements, gait difficulty, and incoordination representing a possible neuromuscular disease of unknown etiology. Although not formally diagnosed in this patient, this could be compatible with the entity described as “LHON-plus” in which LHON patients have systemic neurological involvement.Reference Nikoskelainen, Marttila and Huoponen26 Patient 12 had viral meningitis as an infant with secondary hemiplegia, as well as a fibrosarcoma removed at a young age. Patient 13 had coexistent diagnoses of lupus, rheumatoid arthritis, and polycystic ovarian syndrome. There is no known correlation in the literature between these diseases and LHON, and neither of these two patients had a definitive diagnosis of LHON. The remainder of the patients were either healthy or experienced milder nonspecific systemic symptoms.
Limitations of this study include the retrospective nature of the data collection and the small sample size of patients with full clinical information available for review. For patients found to have possible novel mutations, without a full chart review, it is impossible to know whether the mutation is clinically significant, an incidental finding, or related to a different disease. For this reason, the correlation between laboratory findings and clinical findings remains incomplete in regard to these possible novel mutations. Mutations of low homoplasmy in particular are less likely to represent LHON disease, but were included here for completeness. The incidence of 1 in 77,000 found in this study is lower than previously reported,Reference Carelli, Carbonelli and de Coo2 even when all possible MT-ND1, -ND4, and -ND6 mutations were included. Although LHSC is the reference laboratory for the health region, it is conceivable that patients had testing completed elsewhere, and would therefore be missed in our calculation of incidence. Alternatively, this could reflect a lower incidence of LHON in the Canadian population, a possibility which requires further investigation to confirm. Conversely, the LHSC laboratory also receives samples from outside its catchment area of Southwestern Ontario, and therefore these results may not be specifically representative of the region. This study did not analyze other genes more rarely implicated in LHON, such as mitochondrial cytochrome B and ATP6, due to limited availability of comprehensive chart data. Future studies with access to clinical data regarding all implicated mutations are essential for clearly outlining the clinical picture of LHON in the context of both classic mutations and possible novel mutations. If these novel mutations can be identified in other patients with an appropriate clinical picture, this would help confirm their role in LHON.
Currently, there is scant literature documenting LHON in Canada, and there are no previously reported studies describing LHON in Southwestern Ontario. The data from this study contribute to a growing body of literature on this rare disease, helping to further our understanding of its natural history. Overall, the data collected in this study are consistent with that previously reported about LHON, with a few demographic exceptions. The higher proportion of m.14484 mutations may be accounted for by a founder effect of French Canadians migrating across Ontario. Higher proportions of affected females and older patients may actually be reflective of the true LHON patient population, as suggested by other larger studies.Reference Poincenot, Pearson and Karanjia11 Finally, data from this study add to the literature on possible disease-causing mutations for LHON. Given our findings, clinicians should test for LHON mutations in patients outside of the historical demographic bracket and should take care to consider mutations reported to be of unknown significance as potential pathogenic mutations.
Conflicts of Interest
The authors do not have any conflicts of interest or funding sources to disclose.
Statement of Authorship
HM performed data collection and chart reviews, data analysis, and wrote the manuscript.
BS acquired the genetics data and reviewed the manuscript in detail.
CS created the study protocol and ethics submission and reviewed the manuscript in detail.
LB submitted the protocol for local ethics review, performed data analysis, and wrote the manuscript.





 Open access
Open access