


1. Introduction
Because of its specificities and the large variation of its seasonal extent (18-28 × 106km2), sea ice influences the global climate system through a suite of large-scale processes. Sea ice controls and is controlled by the albedo and the fluxes of heat and moisture across the ocean/ atmosphere interface. The sea-ice extent exerts control over thermohaline circulation through the production of deep water during its formation and the stratification of surface waters during the melt period (Reference Dieckmann, Hellmer, Thomas and DieckmannDieckmann and Hellmer, 2010).
Each year, 7Pg of carbon are released into the atmosphere as carbon dioxide (CO2) by human activities. Oceans play a major role in the context of rising atmospheric CO2 levels because 29% of anthropogenic CO2 is taken up by the ocean through physical and biological processes (Reference TakahashiTakahashi and others, 2009). Until recently, sea ice was assumed to be an impermeable and inert barrier to air-sea gas exchange (e.g. for CO2), so global climate models do not include CO2 exchanges between sea ice and the atmosphere (Reference Tison, Haas, Gowing, Sleewaegen and BernardTison and others, 2002;Reference Bates and MathisBates and Mathis, 2009). However, there is growing evidence that sea ice exchanges CO2 with the atmosphere. Reference Golden, Eicken, Heaton, Miner, Pringle and ZhuGolden and others (2007) showed that the columnar sea-ice permeability for liquids drops by approximately two orders of magnitude below a 5% relative brine volume, which corresponds roughly to -5°C for a bulk salinity of 5 (known as the ‘law of fives’). Recent field observations also question the impermeability of sea ice because large CO2 fluxes have been reported over sea ice during winter (Reference HeineschHeinesch and others, 2009; Reference MillerMiller and others, 2011) and spring (Reference Semiletov, Makshtas, Akasofu and AndreasSemiletov and others, 2004; Reference DelilleDelille, 2006; Reference Zemmelink, Delille, Tison, Hintsa, Houghton and DaceyZemmelink and others, 2006;Reference Nomura, Eicken, Gradinger and ShirasawaNomura and others, 2010a, Reference Nomura, Yoshikawa-Inoue, Toyota and Shirasawab; Reference Papakyriakou and MillerPapakyriakou and Miller, 2011). These fluxes must be partly driven by the gradient of the CO2 partial pressure (pCO2) between sea ice and the atmosphere (Reference DelilleDelille, 2006;Reference Nomura, Eicken, Gradinger and ShirasawaNomura and others, 2010a), which indicates the need for further investigations of pCO2 dynamics within sea ice. To date, very few studies have been performed on the dynamics of the carbonate system within natural sea ice. These studies have generally focused on investigating the potential precipitation of carbonate minerals using measurements of pH, total alkalinity (TA) and dissolved inorganic carbon (DIC), rather than pCO2 directly (Reference Gleitz, Van der Loeff, Thomas, Dieckmann and MilleroGleitz and others, 1995; Reference Papadimitriou, Kennedy, Kattner, Dieckmann and ThomasPapadimitriou and others, 2004, Reference Papadimitriou2007; Reference Delille, Jourdain, Borges, Tison and DelilleDelille and others, 2007;Reference Rysgaard, Glud, Sejr, Bendtsen and ChristensenRysgaard and others, 2007;Reference Nomura, Eicken, Gradinger and ShirasawaNomura and others, 2010a). Reference Delille, Jourdain, Borges, Tison and DelilleDelille and others (2007) presented indirect pCO2 values derived from TA and pH. Such computations rely on the validity of the four important equilibrium constants of the aqueous carbonate system, namely the first (K1 ) and second (K 2) dissociation constants for carbonic acid, the Henry’s law constant for CO2 (K H) and the dissociation constant for water (K W) that are estimated only above −1.6°C. Reference MarionMarion (2001) showed that measurements of carbonate mineral solubilities for subzero temperatures (down to −21.6°C) fit with the predictions derived from the four equilibrium constants of the aqueous carbonate system (i.e. K 1, K 2, K h, K w). This suggests that thermodynamic constants relevant to the carbonate system can be assumed valid at subzero temperatures, but these constants have not been carefully, accurately and independently tested at subzero temperatures.
Reference DelilleDelille (2006) and Reference Delille, Jourdain, Borges, Tison and DelilleDelille and others (2007) have presented direct in situ measurements of brine pCO2 in sea ice. Their method is used in the current work to assess the validity of the new method for measuring bulk ice pCO2. Direct in situ measurements of brine pCO2 were performed using the same principle as the equilibration method used for oceanic underway pCO2 measurements (Reference Dickson and GoyetDickson and Goyet, 1994). This method is based on employing an infrared gas analyzer (Licor®) and a silicon membrane equilibrator. Sea water flows continuously through the equilibrator, where it equilibrates with a closed loop of air. A CO2 analyzer monitors the pCO2 of the air in the closed loop. These measurements do not rely on assessment of the dissociation constants of the carbonate system (except for a correction due to temperature changes between the inlet of the system and the equilibrator). However, although this method provides accurate and reliable measurements, it applies only to brine and has three potential drawbacks: (1) the impossibility of accurately tracking the origin of the brine filling the sackholes, (2) poor vertical resolution and (3) the difficulty of comparing measurements of dissolved compounds (e.g. inorganic carbon) with particulate compounds (e.g. microalgae) that are trapped or fixed in the ice matrix. In particular, when studying inorganic carbon dynamics within sea ice, pCO2 at the interface with the atmosphere is of crucial importance because it determines the direction and magnitude of air/sea-ice CO2 transfer. Unfortunately, due to the poor vertical resolution of the measurements, brine cannot provide an accurate estimate of sea-ice pCO2 at the air/sea-ice interface with the atmosphere (e.g. in the top 5 cm).
Reference MillerMiller and others (2011) developed a new in situ method based on silicone exchange chambers, namely, the peepers. Holes are drilled into the ice cover, the peepers are frozen inside at different depths and are supposed to reach equilibrium with their surroundings, even at low temperature. Gas samples are drawn from the peeper with a syringe, then injected into a gas chromatograph (GC). As noticed by Reference MillerMiller and others (2011), there are some discrepancies between the pCO2 derived from the peepers and indirect pCO2 derived from TA and DIC measurements. In addition, this method requires several hours of work in the field. It requires that the peepers be left in the field for several days to reach equilibrium. Thus the method is not adapted for short-time ship-based ice stations. The method currently has a vertical resolution of 20 cm.
The first measurements of CO2 concentration in bulk sea ice were performed by Reference Matsuo and MiyakeMatsuo and Miyake (1966), who found values ranging from 400 to 24 000pLL-1. The gas concentration was measured using the melting-refreezing method coupled with Toepler pump extraction. The gas was analyzed using a mass spectrometer. When the CO2 concentration was >10 000 µLL-1, the sample was analyzed by the commonly applied mass spectrometric method. When it was <10 000µLL-1, the isotope dilution method was used.
Reference Killawee, Fairchild, Tison, Janssens and LorrainKillawee and others (1998) observed a CO2 concentration of ~50 000µLL-1 in artificial calcium carbonate (CaCO3)- saturated freshwater ice, which is on the same order of magnitude as the concentration measured by Reference Tison, Haas, Gowing, Sleewaegen and BernardTison and others (2002) in an artificial sea-ice tank experiment. The gas composition measurements by Reference Killawee, Fairchild, Tison, Janssens and LorrainKillawee and others (1998) and Reference Tison, Haas, Gowing, Sleewaegen and BernardTison and others (2002) were performed in similar ways. They applied the dry-extraction technique previously employed for glacial ice and described by Reference Raynaud, Delmas, Ascencio and LegrandRaynaud and others (1982) and Reference Barnola, Raynaud, Neftel and OeschgerBarnola and others (1983). Gas samples were crushed at very low temperature (−55°C) under vacuum (1.333 Pa), so that the technique measured the gas composition both from gas bubbles and from gas dissolved in the sea-ice brine. An extraction line was used in conjunction with a Varian 3300® Gas Chromatograph.
Reference Tison, Haas, Gowing, Sleewaegen and BernardTison and others (2002) suggested that two major biases could explain the high CO2 concentrations measured in their sea-ice tank experiments. First, the CaCO3 precipitation observed during the experiment could have promoted a degassing of CO2, according to
Second, uncontrolled sustained bacterial activity could also act as a CO2 source. However, more recent work has suggested a potential methodological bias in these types of measurements (Reference VerbekeVerbeke, 2005). Since they were obtained using gas extraction under vacuum at low temperature, the carbonate system was significantly disturbed, leading to the release of CO2 in the headspace in larger quantities than those that were effectively initially dissolved within the brine. In sea water, as well as in brine, most CO2 is dissociated in carbonates (CO3 2-) and bicarbonates (HCO3 -) (Reference Zeebe and Wolf-GladrowZeebe and Wolf-Gladrow, 2001). Brine pCO2 depends on the equilibrium between CO2, HCO3 - and CO3 2-, which is governed by the dissociation constants of the carbonate system (K1 , K 2):
These constants are strongly affected by changes in salinity and temperature (Reference Zeebe and Wolf-GladrowZeebe and Wolf-Gladrow, 2001). According to Reference Papadimitriou, Kennedy, Kattner, Dieckmann and ThomasPapadimitriou and others (2004), a decrease in temperature promotes an increase in the brine concentration and, consequently, an increase in the brine pCO2. Additionally, the vacuum in the vessel forces the transfer of CO2 from the liquid phase to the headspace. In turn, removal of CO2 forces the displacement of the equilibrium so that the CO2 removed from the liquid phase is rapidly replaced by CO2 converted from the pool of CO3 2- and HCO3 -. As a result, measuring the CO2 concentration in the headspace of the vessel leads to a value that is not related to the in situ pCO2 or DIC because the total extraction of DIC cannot be achieved without acidification. Overall, the CO2 concentration in the headspace under a vacuum is useless in constraining the carbonate system.
The common method for measuring the pCO2 of a liquid phase is to measure the concentration in a headspace in equilibrium with the liquid phase at a pressure close to atmospheric pressure. We applied this rationale to sea ice and developed a method for determining the pCO2 of bulk sea ice that can be performed with a vertical resolution of 5 cm. This resolution allows accurate assessment of the pCO2 gradients in the ice and at the air/sea-ice interface and better comparisons of pCO2 with crucial biogeochemical parameters. This method requires relatively little fieldwork. Only one core needs to be collected in addition to the measurement of the temperature and salinity profile. Measurements were performed in standard artificial sea-ice samples synthesized in a laboratory with reproducible properties to test the accuracy and reproducibility of the method. The method was then validated against direct in situ brine pCO2 measurements performed according to the method proposed by Reference Delille, Jourdain, Borges, Tison and DelilleDelille and others (2007).
2. Materials and Methods
The method used in this study was first tested on standard sea ice with homogeneous properties (e.g. salinity, texture and pCO2 content), to assess the method’s reproducibility and the precision of the measurements.
We also tested the method on natural sea-ice samples by producing high-resolution profiles and comparing them with in situ brine pCO2 measurements. Samples were collected at the same location (i.e. within an area of 25 m × 25 m) on first- year landfast sea ice offshore of Niksiuraq at the end of the road to Point Barrow, Alaska (71.37055° N, 156.51363° W) on 10 April, 12 May and 5 June 2009 (denoted as stations 7, 9 and 10 respectively).
The method was then used on artificial sea ice produced in the frame of the Interice IV sea-ice mesocosm experiment carried out in the Hamburg Ship Model Basin (HSVA) ice tank. Sea ice was formed in a 1 m3 polyethylene bag filled with North Sea water. Atmospheric temperature was about −15°C. Bags were equipped with PVC tubing to allow for water-pressure equilibration during ice growth. Water underlying the enclosure was mixed continuously with an underwater pump.
2.1. General principle of the pCO2 analytical procedure
The method described below was inspired by Reference WeissWeiss (1981), Reference Dickson and GoyetDickson and Goyet (1994) and Reference Neill, Johnson, Lewis and WallaceNeill and others (1997), who proposed procedures to measure oceanic pCO2 in discrete water samples. The general principle of our method is as follows: a sea-ice sample of known volume is equilibrated at the observed in situ temperature with a mixture of nitrogen and CO2 of known concentration (the so-called standard gas); once the ice sample is equilibrated, the air phase is injected into a Varian 3300® Gas Chromatograph, to measure the CO2 concentration.
2.2. Synthesis of standard artificial sea ice
Reference Jacka and LileJacka and Lile (1984) proposed a method for creating artificial freshwater ice by mixing fresh water at the freezing point with individual ice crystals. The principle underlying this technique is simple. The freshwater-ice crystal mix is compressed under a mechanical press. As the pressure increases, the freezing point of the water decreases and the liquid water is expelled from the container. Upon sudden release of the pressure, the freezing point of the water increases and allows bulk freezing of the mixture. According to Reference Jacka and LileJacka and Lile (1984), this method is reproducible and produces ice samples with constant physical-chemical properties. Moreover, the ice crystals comprising this kind of ice do not show preferential orientation.
In this study, this technique was adapted for sea-ice production. Sea water (deep sea water from the Bay of Biscay) was stored in an ice-cream maker in a -20°C cold room to bring it to its freezing point. By mixing the newly formed slush the continuous rotation of the blades of the ice-cream maker avoided formation of large sea-ice crystals and prevented salt segregation due to rejection of impurities (Reference Weeks and AckleyWeeks and Ackley, 1982). When the slush density became high enough to hamper the rotation of the blades, the slush was transferred to a dedicated vessel and squeezed to a pressure of ~2 × 106Pa; the pressure was then released. Directly after sea-ice formation, the block was stored in a freezer at -30°C for at least 48 hours before further treatment.
The sea water used to produce this standard sea ice was aged sea water for the purpose of limiting CO2 production from biological activity. According to Reference Takahashi, Olafsson, Goddard, Chipman and SutherlandTakahashi and others (1993), a decrease of 1 °C acts to reduce pCO2 by ~4.1%. To maintain a constant and known value of the pCO2 of the sea water before ice formation, despite the decrease in temperature, air with an atmospheric CO2 concentration (~385 µLL-1) was bubbled continuously in the solution for 24 hours prior to ice formation and subsequently throughout the process of standard sea-ice formation. The bubbled air was cooled down using a cooling coil before being injected into the sea-water reservoir.
To assess the homogeneity of the properties of the standard sea ice, a set of salinity profiles and examination of ice thin sections were performed.
2.2.1. Salinity of artificial ice
The ice sample must have a rectangular parallelepiped shape of 4 cm × 4 cm × 4.4 cm to fit tightly in the analysis vessel. A sample was cut from the center of the standard sea-ice block to avoid contamination from the squeezed salty water or from brine rejection during storage of the ice at −30°C. It was then cut into slices ~1 cm thick (slice 1, 2,..., n), and each slice was cut into four identical pieces (A-D). The ice was melted in a closed receptacle at ambient temperature, and salinity was measured using a TitraLab TIM 870® with a cell of conductimetry CDC 565 from an Analytical® radiometer. A Heto® thermostatic bath was used to maintain a constant temperature of 25°C, with a precision of ~±0.1°C. The constant of the cell of conductimetry was calibrated before each set of analyses using a standard Merck® KCl solution at 0.01 mol L-1 with known conductivity at 25°C.
2.2.2. Thin sections
Vertical and horizontal thin sections were produced following standard procedures (Reference TisonTison and others, 2008) to describe the texture of the ice. Images of the texture of the crystals were collected from the thin sections using a light table and cross- and parallel-polarized sheets with a macro setting on the camera (Nikon® Coolpix S200, 7.1 megapixels).
2.3. Sea-ice sampling
Sea ice from the Interice IV experiment was collected by sawing a 10 cm × 20 cm block of ice according to the method of Reference Tison, Haas, Gowing, Sleewaegen and BernardTison and others (2002), then wrapped in polyethylene (PE) bags and stored at −30°C.
An electro-polished stainless-steel corer with an internal diameter of 14 cm was used to retrieve natural ice cores at Barrow. Cores were immediately wrapped in PE bags and stored on the sampling site in an insulated box filled with individual cooling bags precooled at−70°C. This precaution served to minimize brine drainage from the samples (Reference TisonTison and others, 2008).
The sea-ice temperature was measured in situ directly after the extraction of the core using a calibrated probe (TESTO 720®) inserted into pre-drilled holes (~3 cm depth) perpendicular to the sides of the core. The diameter of the hole matched that of the temperature probe. The measurement precision was ±0.1 °C. This ‘temperature core’ was cut into successive, 5 cm thick slices in the laboratory. Each slice was stored in a bucket at 4°C in the dark to melt it, and the salinity was measured with a Thermo-Orion® portable salinometer WP-84TPS with a precision of ±0.1.
To measure the TA on bulk sea ice, we followed the same procedure as Reference MillerMiller and others (2011). An ice sample was cut in the laboratory, and placed in a gas-tight laminated NEN/PE plastic bag fitted with a gas-tight Tygon tube and valve for sampling. A solution of supersaturated HgCl2 was added to the plastic bag to halt biological activity. The bag was immediately closed, and excess air was removed through the tube using a manual air pump. The ice was allowed to completely melt at +4°C in the dark, then TA analysis was immediately undertaken. The maximum potential leakage has been estimated as <20 mmol kg-1 (Reference MillerMiller and others, 2011). TA was measured on brine and on bulk sea ice by open-cell titration with HCl 0.1 mol L-1 (Reference GranGran, 1952) on 50 mL of sea-ice meltwater samples. Titration was stopped for 10 min at pH 4.2 to ensure that all CaCO3 crystals were dissolved, prior to TA measurement over the pH range 3-4.2, required for the Gran function. The accuracy of TA measurements was ±4 mmol kg-1. Data were quality- checked with certified reference material acquired from Andrew Dickson (Scripps Institution of Oceanography, University of California, San Diego).
2.4. Ice pCO2
2.4.1. Dedicated container for pCO2 measurements in sea-ice samples
A special sample container was designed to perform pCO2 measurements in the sea-ice samples. The ice sample was required to fit precisely into this container to minimize the headspace. The container also had to be thermally conductible to maintain the ice sample temperature during the equilibration procedure at the field in situ temperature. The container was made of epoxy resin, which is inert and does not interact with CO2. This resin was mixed with an aluminum powder at a ratio of 20% aluminum to 80% resin, to improve heat conduction across the container walls (Fig. 1).

Fig. 1. Dedicated container for the measurement of pCO2 in solid sea-ice samples, equipped with a flat glass top, silicon joint and a clamp. The black and white line represents the centimetric scale.
The container was equipped with a flat glass top including a tap and a silicon joint to ensure airtightness. This top was designed to reduce the headspace above the ice sample. Several tests showed that the container could absorb a small portion of the standard gas. Therefore the container was stored filled with the standard gas at a pressure higher than atmospheric pressure to saturate the container adsorption sites with CO2.
2.4.2. Vacuum line and gas chromatograph
An extraction line (Fig. 2) was used in conjunction with a Varian 3300® Gas Chromatograph. The latter device is equipped with a Haysep column and a flame ionization detector following catalytic transformation into methane. It allows the measurement of oxygen and argon (as a single peak), nitrogen, methane and CO2. The carrier gas is helium. The extraction line is made of stainless steel. It is equipped with a vacuum pump and a Pfeiffer® pressure indicator (with optimized sensitivity in the atmospheric range). A water trap consisting of a mixture of ethanol and liquid nitrogen at −65°C is placed just before the inlet of the GC. pCO2 is therefore measured in dry air because the water vapor is trapped before injection.

Fig. 2. Sketch of the different components forming the extraction line, including the Varian 3300® Gas Chromatograph.
The precision of the measurements made with these system settings is ±5% (Reference Tison, Haas, Gowing, Sleewaegen and BernardTison and others, 2002). Accordingly, a mean relative error of 5.03% was obtained from 30 injections of standard gas ([CO2]=298µLL-1), with a standard deviation of 17.15 µLL-1 and a variation coefficient (CV) of 5.9%.
2.4.3. Temperature equilibrium
Temperature is a crucial parameter for pCO2 analyses, as pCO2 is strongly related to temperature. Additionally, temperature controls brine salinity (Reference Weeks and AckleyWeeks and Ackley, 1982;Reference Eicken, Thomas and DieckmannEicken, 2003) and thus pCO2 (Reference DelilleDelille, 2006; Reference Delille, Jourdain, Borges, Tison and DelilleDelille and others, 2007). Finally, temperature controls brine volume and, consequently, permeability, which is a critical parameter for our purposes because equilibrium can be achieved only if the ice is permeable to gas. Therefore, the bulk ice pCO2 must be measured at the in situ temperature (i.e. the sea-ice temperature measured in the field during core sampling). As the ice samples were stored at -30°C, it was necessary to warm the sample to the in situ temperature during the equilibration procedure between the ice sample and the standard gas. The ice sample within the container was therefore stored in a thermostatically controlled bath during the equilibration.
According to Reference Eicken, Thomas and DieckmannEicken (2003) and references therein, the thermal conductivity of sea ice depends on the temperature gradient and the sea-ice salinity. The specific heat capacity of sea ice, C si, is expressed as
where C i = 2.11 Jg-1 K-1 is the specific heat capacity for pure ice, Si is the ice salinity and T is the temperature (°C). According to this equation, more energy is needed to warm a sea-ice cover sample by 1°C at a temperature close to the freezing point of sea water than at a lower temperature. Therefore, to reach the in situ temperature more rapidly, the thermostatic bath was first set to −2°C. Once the ice sample had reached the in situ temperature, the bath temperature was adjusted to the in situ temperature for fine temperature adjustment. Tests were performed on artificial sea ice to determine the time required to equilibrate the temperature of the ice with the temperature of the thermostatically controlled bath. Tests were performed on ultrapure water with NaCl added to obtain salinities of 4, 6, 8, 10, 16 and 23. The solution was frozen and stored at −30°C. Once frozen, the ice was cut, adjusted to the pCO2 container inner volume, and a temperature probe (a calibrated Testo 720® with a precision of ±0.2°C) was inserted into its center. The container with the ice sample was placed into a thin plastic bag and submerged in a thermostatic bath at−2°C. Temperature was recorded every 5 min until an ice temperature of −2 ± 0.5°C was reached. Thus, we determine the time needed, as a function of ice salinity, to reach the desired temperature.
2.4.4. pCO2 analysis
According to Reference Golden, Ackley and LytleGolden and others (1998), because the ice samples were stored at −30°C, their brine volume should be <5%, so they can be considered impermeable to gas exchange with the atmosphere. Therefore, the sea-ice samples could be manipulated in the absence of contamination in the cold room, maintained at −30°C.
The same procedure was followed for both artificial standard and natural sea-ice samples. The day before analysis, the container was filled with standard gas to saturate the potential adsorption sites of its inner surface. Thirty minutes before the beginning of the sample treatment, the container was precooled in the cold room.
In the cold room, the ice sample was cut with a bandsaw and adjusted to the container’s inner volume (4cm × 4 cm × 4.4 cm). The sample was sanded down using finegrained sandpaper so that it fit tightly into the container to both minimize the headspace volume and obtain a constant headspace volume. Next, the container, equipped with its glass top and the silicon joint, was placed into a dewar filled with a mixture of ethanol-liquid nitrogen at −30°C. The container was then connected to the extraction line (tap closed). The line was first evacuated down to a pressure of 1.333 × 10-1 Pa, then the container was evacuated for 5 min (tap open for a maximum of 5 min to avoid drawing the absorbed CO2 from the container due to the vacuum). The standard gas was then injected into the container at 101 300 Pa, and the container was subsequently removed from the extraction line (tap closed), placed in a thin plastic bag and submerged in a thermostatic bath (set to −2°C). The equilibration process was then begun. The ice sample was warmed and equilibrated with the standard gas. Following the salinity of the sample and the appropriate temperature- salinity relationship, 30 min before the sample was expected to reach the in situ temperature the bath temperature was adjusted to the in situ temperature. The sea-ice sample was then left in the thermostatic bath for an additional 90 min to reach the in situ temperature and complete the equilibration. Subsequently, the container was placed in a dewar filled with an ethanol-liquid nitrogen mixture at the in situ temperature and reconnected to the evacuated (1.333 × 10-1 Pa) extraction line. At the same time, a water trap consisting of a dewar filled with an ethanol-liquid-nitrogen mixture at −65°C was placed on the line just before the GC. Finally, the gas was injected into the GC. Immediately after injection was completed, the ice sample temperature was measured using a calibrated temperature probe (Testo 720®).
3. Results and Discussion
3.1. Standard artificial sea-ice properties
3.1.1. Thin sections
In both transversal and longitudinal thin sections, the ice crystals were of millimetric size and did not show any preferential orientation, in good agreement with the findings of Reference Jacka and LileJacka and Lile (1984). These types of crystals and the absence of orientation are typical of granular ice (Reference Tison, Lorrain, Bouzette, Dini, Bondesan, Stievenard and JeffriesTison and others, 1998;Reference Eicken, Thomas and DieckmannEicken, 2003).
3.1.2. Salinity
The salinity profiles of each block of artificial sea ice are shown in Figure 3. The salinity of each section ranged from 7.5 to 23, while the bulk salinities of the blocks were 10.1 − 17. This is significantly higher than the mean salinity of natural sea ice (Reference Nakawo and SinhaNakawo and Sinha, 1981;Reference Weeks and AckleyWeeks and Ackley, 1982;Reference MeeseMeese, 1989;Reference Eicken, Thomas and DieckmannEicken, 2003) due to the growth of this ice in a closed small bath. Brine rejection associated with the sea-ice growth increases the salinity of the residual sea water incorporated into the ice during squeezing. All profiles showed a trend of increasing salinity towards the bottom of the block, suggesting that a consolidation process had occurred moving from top to bottom, with variable salt segregation from block to block.

Fig. 3. Salinity profile in eight artificial standard sea-ice blocks. The dashed line shows the mean salinity of the bulk block while the blacktriangle shows the mean salinity of each slice of the block.
Only block 1 exhibited significant salinity heterogeneity at a given depth. The salinity range of block 1 was the greatest, with salinity ranging from 8.4 to 17.4, while the other blocks were more homogeneous, with a maximal dispersion of the salinity of ±4 at the same depth.
The salinity profiles of this artificial standard sea ice will be affected by several processes. First, the initial grain size should influence the porosity of the medium, but thin sections did not exhibit significant differences with respect to grain size. The pressure applied to squeeze the slush should be constant. Nevertheless, some variations may have occurred due to the imprecision of the pressure indicator. Second, a leak may occasionally occur at the bottom of the container used during pressurizing, and the bottom of this container was not completely watertight if not firmly screwed. Hence, as the pressure increased, most of the residual sea water was expelled at the top of the container, but occasionally some water was also expelled at the bottom. This could explain the range of bulk ice salinities observed. However, each block of standard sea ice was considered reasonably homogeneous.
As a further test, in parallel with the pCO2 tests, a thin slice was cut from the analyzed sea-ice block to measure its bulk salinity. We measured 36 samples;the mean salinity was 16 with a standard deviation of 1.05 (coefficient of variation 6.5%;Table 1). Therefore, two samples cut side by side from the same block of artificial standard sea ice were considered similar in terms of salinity and other physical properties and, thus, in terms of pCO2.
Table 1. Samples A-K were analyzed with a standard gas of 298 pL L-1 while samples M-Y were analyzed with a standard gas of 1483 µLL-1. Tis the temperature measured in the sample after the injection in the GC. S is the salinity measured on a thin slice of ice from the block of standard sea ice. Vb/V was determined from T and S. pCO2 is the concentration measured by the GC

3.2. Time required for temperature adjustment
Warming of the ice was rapid and linear up to -5°C. Above -5°C, the rate of temperature increase decreased steadily, tending asymptotically towards the temperature of the thermostatic bath (-2°C;Fig. 4). For each salinity range, several tests were performed to assess the precision and accuracy of this method of warming the ice to a given temperature.

Fig. 4. Evolution of ice temperature with time as a function of mean ice salinity for artificial ice samples immersed in a bath at –28C. The bold line in each panel is the mean trend. The slight increases in ice temperature observed in the S = 16 and S = 23 panels are due to bad precooling of the container before the beginning of the equilibration.
Using this procedure, the in situ temperature could be reached within ±1°C (Fig. 4), which was not fully satisfactory. To make the temperature adjustment more reliable and precise, 30 min before the sample was expected to reach the in situ temperature (following the temperature-salinity relationship) the temperature of the thermostatic bath was adjusted to the in situ temperature, and the sample was left in the bath for an additional 90 min. The total elapsed time needed to warm the ice sample ranged from 120 to 300 min, depending on the in situ temperature of the ice sample.
3.3. pCO2 measurements in standard sea ice
Sea-ice brine equilibrates with the atmosphere (or the standard gas) following Henry’s law, where K H is the Henry constant:
A prerequisite of this equilibrium is that sea ice must be permeable to gas exchanges. According to Reference Golden, Ackley and LytleGolden and others (1998, Reference Golden, Eicken, Heaton, Miner, Pringle and Zhu2007), sea ice may be considered permeable once its relative brine volume (V b/V, where V b is the brine volume and V the total ice volume) is higher than a threshold of 5%. The relative brine volume was calculated from salinity and temperature data (Reference Eicken, Thomas and DieckmannEicken, 2003) and was found to be >5% for all artificial ice samples (Table 1).
The accuracy and precision of this method were estimated by measuring the CO2 concentration in twin samples of artificial standard sea ice. These measurements were performed at various temperatures and with two different equilibration standard gases with concentrations of 298 and 1483 pLL-1, respectively, and the results are shown in Figure 5. By observing both datasets, a common general trend may be observed. pCO2 appears to be inversely correlated with ice temperature. Using the 298µLL-1 standard gas, pCO2 ranged from 1667 µLL-1 at-15.3°C to 285 pL L-1 at -3.5°C. Down to a temperature of -9.5°C, the results exhibited good reproducibility, with a CV of ~2.9% between twin samples. Below -9.5°C, greater dispersion was observed for one of the experiments, where the CV increased to 17.6%, which is out of the range of precision calculated for the GC measurements (CV = 5.9%). Using the 1483 µLL-1 standard gas, pCO2 ranged from 1775 µLL-1 at −12.7°C to 211 µLL-1 at −1.8°C. Samples P, Q and R were analyzed at different temperatures and therefore presented pCO2 values that were significantly different. In contrast, samples O and S were analyzed at the same temperature and showed good reproducibility, with a CV close to the range of precision of the GC measurements. Samples analyzed below −10°C showed a larger dispersion, with CVs about twice the precision of the GC.

Fig. 5. pCO2 of bulk sea ice using equilibration standard gas of (a) 298µLL–1 and (b) 1483µLL–1. The standard gas concentration is represented as a dashed line. Each couple of points represents twin samples from the same block of standard sea ice. The error bar shows the GC precision (5.9%).
The general trend of decreasing pCO2 as ice temperature increases has previously been described by Reference DelilleDelille (2006). As ice temperature increases, the meltwater from the ice crystals dilutes the brine, so the pCO2 decreases. Associated with the brine dilution, potential precipitates of CaCO3 within sea ice (Reference DieckmannDieckmann and others, 2008, Reference Dieckmann2010) may dissolve, promoting pCO2 decrease. In Figure 6, we compare the results of our artificial standard sea ice to natural sea-ice data from Reference DelilleDelille (2006) and Reference Delille, Jourdain, Borges, Tison and DelilleDelille and others (2007), who measured in situ the pCO2 of sackhole brine. The range of concentrations measured using this new method is consistent with the natural sea-ice dataset. Our results are also in agreement with findings from in situ brine pCO2 measurements sampled in the Arctic at Barrow (data not shown) and during the Circumpolar Flaw Lead (CFL) System Study field program (Reference BarberBarber and others, 2010), in which in situ brine pCO2 values were measured from April to June 2008 following the same procedure as Reference Delille, Jourdain, Borges, Tison and DelilleDelille and others (2007) and Geilfus and others (in press). It should be noted that when comparing the artificial sea-ice pCO2 values obtained with the two different standards, it was observed that the pCO2 values equilibrated with the 298pLL-1 standard were systematically lower than those measured with the 1483µLL-1 standard, suggesting a potential bias due to the choice of standard used for equilibration. The number of moles exchanged between the standard gas and the brine medium must be negligible compared to the number of moles initially present within the brine to ensure that the equilibration procedure between the ice sample and the standard gas does not affect the initial brine pCO2. To reduce the number of moles exchanged between the standard gas and the brine medium, the pCO2 of the standard gas should be close to the expected pCO2 of brine.

Fig. 6. Comparison between pCO2 measurements on bulk standard sea ice and in situ measurements of brine pCO2.
3.4. Influence of the headspace-volume/ice-volume ratio
One of the potential errors of the above method comes from the amount of CO2 exchanged between the standard gas and the brine medium that cannot be readily estimated. Hence, the number of moles exchanged between the standard gas and the brine medium must be negligible compared to the number of moles initially present within the brine. This requires that the volume of brine be sufficiently large compared with the volume of the standard gas and that the concentration of the standard gas used for the measurements be as close as possible to the expected concentration of the ice sample. Reference Dickson and GoyetDickson and Goyet (1994) suggest an air/sea- water volume ratio of 0.087 to achieve a precision of 2.5%. In the procedure applied in the present study, the sample size was 4 cm × 4 cm × 4.4 cm (70.4 mL). The internal volume of the container equipped with the glass top and silicon joint was 83.39 mL. Hence, the air-volume/ice-volume ratio was 0.18. However, only the brine equilibrated with the headspace. Considering a minimum relative brine volume of 5% (permeability threshold), the brine volume was 3.52 mL. This led to a headspace-volume/brine-volume ratio of 3.69 (12.99 mL/3.52 mL), which should be compared to the ratio of 0.087 advocated by Reference Dickson and GoyetDickson and Goyet (1994). However, it should be noted that the DIC of brine can be significantly higher than the DIC of sea water (Reference Rysgaard, Glud, Sejr, Bendtsen and ChristensenRysgaard and others, 2007, Reference Rysgaard, Bendtsen, Pedersen, Ramlov and Glud2009;Reference MillerMiller and others, 2011) as a result of brine concentration, so the air-volume/liquid-volume ratio for sea water and brine cannot be readily compared. The existence of a high air-volume/brine-volume ratio emphasizes the fundamental need for specific dedicated tests to ensure the accuracy and reproducibility of the method.
Brine volume depends on temperature, salinity and ice volume. To decrease the headspace-volume/brine-volume ratio, it is necessary to decrease the headspace-volume/ice- volume ratio as much as possible. We attempted to assess the bias related to various headspace-volume/ice-volume ratios and various differences between standard gases and the pCO2 values of brine with a simple model (Fig. 7). The model iteratively computes the amount of CO2 exchanged between brine at a given pCO2 (pCO2;,-) and the headspace with a given standard to reach an equilibrium, taking into account TA of the brine and the CO2 dissociation coefficients. CO2 speciation was calculated using numerical routines for the calculation of carbonate system parameters from Reference Zeebe and Wolf-GladrowZeebe and Wolf-Gladrow (2001), the CO2 acidity constants of Reference Mehrbach, Culberson, Hawley and PytkowiczMehrbach and others (1973) refitted by Reference Dickson and MilleroDickson and Millero (1987), the CO2 solubility coefficient of Reference WeissWeiss (1974), the SO4 2- dissociation constant of Reference DicksonDickson (1990a) and the borate acidity constant of Reference DicksonDickson (1990b), while the total borate molality was calculated using the Reference UppstrdmUppstrom (1974) ratio to chlorinity. We assumed conservative behavior of the CO2 dissociation constants at subzero temperatures. Reference MarionMarion (2001) and Reference Delille, Jourdain, Borges, Tison and DelilleDelille and others (2007) suggested that thermodynamic constants relevant to the carbonate system can be assumed to be valid at subzero temperatures. The difference between the pCO2 at equilibrium and pCO2,- is the measurement error due to the exchange of CO2 between the sample and the headspace. The error increases significantly both when the chosen standard shows an increasing difference from the real value and when the headspace- volume/ice-volume ratio increases. It should also be noted that the results of these calculations suggest there are no geometrical constraints on the equilibrium process.

Fig. 7. Error in the method following different V air/V ice ratios and different concentration of standard gas. The initial conditions at the computation were T=−5ºC, S = 6, TA= 2400 µmol kg–1 and brine pCO2 = 400µLL–1 (a) and 750µLL–1 (b).
3.5. pCO2 measurement in Arctic sea ice
The above method was then applied in natural sea-ice samples to produce high-resolution profiles and to compare them with in situ brine pCO2 measurements to assess the accuracy of the method. Ensuring that sea ice is permeable to gas exchange is a prerequisite to apply this new method and to measure the pCO2 of bulk ice under in situ conditions. Therefore, the brine volume was calculated from temperature and salinity data collected in the field according to the formula of Reference Eicken, Thomas and DieckmannEicken (2003).
The brine volume was generally <5% until reaching station 8, where the 5% threshold was exceeded. Therefore, bulkpCO2 measurements were first applied at station 9. Here the larger diameter of the ice core provided the opportunity to measure twin samples, allowing us to check the reproducibility and precision for natural sea-ice samples. Station 10 was then analyzed to compare the results from this method to direct in situ measurements of the brine pCO2 available at that station. These data were obtained following the protocol of Reference Delille, Jourdain, Borges, Tison and DelilleDelille and others (2007). This provided a test of the accuracy of the method. Finally, station 7 was analyzed. This station presented a brine volume below the permeability threshold, and the limits of the method were tested in this respect. All the samples were analyzed using a 318mLL-1 standard gas. The top 20 cm of these stations exhibited a frazil sea-ice texture made of ice crystals ranging from a few mm to 1 cm. The rest of the ice column was columnar sea ice, with large centimetric crystals, vertically elongated.
The pCO2 profile at station 9 was undersaturated with respect to the atmospheric concentration (393 mLL-1, GLOBALVIEW-CO2, 2010; http://www.esrl.noaa.gov/gmd/ccgg/globalview/co2/co2_intro.html) except between 10 and 30cm depth, with concentrations ranging from 40 to 442µLL-1(Fig. 8). A first pCO2 minimum was observed at 5 cm depth, with a pCO2 of ~103µLL-1. Below this value, pCO2 increased to its maximum concentration at 25cm depth. From 30 to 110cm depth, pCO2 increased slightly from 250 to 350µLL-1. It then decreased to 40µLL-1 at the bottom of the ice core. Profiles from replicate ice samples exhibited the same general trend, except for three values at 50, 70 and 80 cm depth.

Fig. 8. High-resolution pCO2 profiles on natural sea ice sampled in Barrow. Stations 7, 9 and 10 are shown. The dashed line shows the standard gas concentration (318µLL–1) while the dotted line shows the atmospheric CO2 concentration. The in situ measurements of brine pCO2 are plotted as white stars. The gray area in station 7 is the impermeable part of the ice, as determined from the relative brine volume threshold of 5%, according to Reference Golden, Ackley and LytleGolden and others (1998, Reference Golden, Eicken, Heaton, Miner, Pringle and Zhu2007).
The pCO2 profile at station 10 was undersaturated throughout the thickness of the ice sample, with concentrations ranging from 23 to 240µLL-1. pCO2 at the ice interface with the atmosphere was 61µLL-1. It increased slightly to 160µLL-1 at 20cm depth, then decreased to 22µLL-1 at 30 cm depth. Below 30 cm, the general trend of the profile was an increase in pCO2 with increasing ice depth, up to 193µL L-1 at the bottom of the ice sample. This profile can be compared to the in situ measurements of brine pCO2, which ranged from 26 to 74µLL-1. Both measurements were on the same order of magnitude, although the in situ measurements presented slightly lower values than the measurements in bulk sea ice.
The pCO2 at station 7 ranged from 40 to 327 µLL-1. The top 90cm of the ice exhibited a brine volume below the permeability threshold. In this region, with a few exceptions, the pCO2 was stable, presenting values around the concentration of the standard gas (318µLL-1). Below the depth corresponding to the permeability threshold, the pCO2 decreased from 327µLL-1 to 40µLL-1 at the bottom of the ice sample. In situ measurements of brine pCO2 were also performed, with values ranging from 1352 to 1063µLL-1. These values were significantly higher than the results for pCO2 in bulk sea ice. This apparent discrepancy can be explained by taking into account that sackhole brine collects material from the entire sea-ice cover above the bottom level of the sackhole. Therefore, its pCO2 can easily be biased by the signal of the colder upper layers.
The reproducibility and precision of this method were confirmed by the results from station 9. The same general trend as in station 7 was observed, and a CV of 8.8% was calculated. This was slightly higher than the CV of the GC measurements (5.9%). According to Reference Eicken, Lange and DieckmannEicken and others (1991), however, sea ice is a very heterogeneous medium, even at a high resolution, depending on the brine channel location. Therefore, considering the difficulty of ensuring homogeneous samples within sea ice, we consider this range of reproducibility to be acceptable.
The comparison between the pCO2 measured in the bulk ice and the in situ brine pCO2 is a measure of the accuracy of our new method. Brine samples were collected in sackholes (Reference Gleitz, Van der Loeff, Thomas, Dieckmann and MilleroGleitz and others, 1995). This is currently considered to be the best available method to sample brine for chemical studies (Reference Papadimitriou, Kennedy, Kattner, Dieckmann and ThomasPapadimitriou and others, 2004). However, as noted above, sackholes do not enable us to track the origin of the brine, and they provide a record that could be biased due to a dominant input from a given level into the sea-ice cover. This characteristic may explain the moderate difference observed between the two methods. Another possibility is the potential bias towards higher pCO2 values resulting from the choice of the standard at 318µLL-1. However, the high-resolution profile still presents features inherited from the previous profile of station 9, such as the relatively broad minimum between 30 and 80 cm and the relatively broad maximum between 90 and 130cm. At this stage, the generally good agreement between the in situ brine measurements and the pCO2 determined in bulk sea ice suggests that this new method is reasonably accurate.
Using the bulk ice salinity, the ice temperature and TA of the brine at the four sackhole levels of station 10, it was possible to assess the error in the pCO2 values measured with our new method as a function of the V atm/V ice ratio and for various equilibration standards used. This is plotted in Figure 9. Note that the carbonate equilibrium constants used for these calculations are only strictly valid for higher temperatures and generally lower salinity conditions.

Fig. 9. Error estimated following the chemical conditions measured in brine from station 10. Br 1: T = –0.08ºC, S = 2.7, pCO2 = 73, TA= 319 µmol kg–1. Br 2: T = –0.29ºC, S = 5.1, pCO2 = 28, TA= 474 µmol kg–1. Br 3: T = –0.54ºC, S = 3.4, pCO2 = 26.1, TA= 344 µmol kg–1. Br 4: T = –0.09ºC, S = 2.3, pCO2 = 34.4, TA = 276µmol kg–1. The dashed line shows the V air/V ice ratio from this method; the dotted line shows the precision of the GC.
Using the V atm/V ice ratio in our settings and the concentration of the standard gas (300mLL-1), the error due to the injection of a standard gas with a given CO2 concentration in the sample ranged from 2 mLL-1 (Br 3) to 8 mLL-1 (Br 1). This error increases dramatically when a standard gas with a higher concentration is used, while the inverse is true when the chosen standard gas has a concentration closer to that of the brine pCO2. In our case, the use of a standard gas with a CO2 concentration of 150µLL-1 would have provided an error less than the precision of the GC measurements. Therefore, it is essential to make a first assessment of the expected pCO2 levels, either by measuring the brine pCO2 in situ or using the brine-pCO2-ice-temperature relationship reported by Reference DelilleDelille (2006).
The measurement of the pCO2 of bulk sea ice at station 7 demonstrated the limitation of our method when the ice is impermeable. The equilibration process between the standard gas and the ice sample is hampered in such cases, resulting in measured pCO2 values close to that of the standard gas used. This was confirmed by the concentrations obtained in the impermeable portion of the ice from station 7. As the ice became permeable in the lower portion of station 7, coherent changes in pCO2 were again detected. The detailed profiles in Figure 8 show the need for further analysis of the processes controlling pCO2 dynamics. This will be provided in a dedicated paper with the assistance of the full suite of ancillary physical and biogeochemical parameters collected during the Barrow 2009 experiments.
3.6. pCO2 measurements on Interice IV sea ice
Our method has also been tested on sea-ice samples collected in the framework of the Interice IV experiment (Reference Thomas, Grune and BretelerThomas and others, 2010). At the end of the experiment, the thickness of this artificial sea ice grown in the HSVA experimental tank ranged from 17 to 21 cm, with 1 cm of frazil ice at the top while the rest of the ice cover was columnar ice (Fig. 10). According to the temperature and salinity measured regularly during the sampling period, brine volume was generally >5% throughout the ice thickness, with the exception of a few episodes slightly below Golden and others’ (1998) critical value, at depths of 0-15 cm (data not shown).

Fig. 10. Thin section of sea ice from bags A and B from Interice IV experiment. The scale is provided in cm.
During sea-ice sampling, the in situ brine pCO2 and seawater pCO2 were measured using the procedure for in situ pCO2 measurement described by Reference Delille, Jourdain, Borges, Tison and DelilleDelille and others (2007). We were thus able to compare results from our new method with in situ measurements. We carried out the analysis using gas standards of different CO2 mixing ratios in order to assess the effect of the choice of the standard pCO2 on the measurement. We first used a standard gas with a pCO2 of 396µLL-1. pCO2 observed in bag A ranged from 42 to 608µLL-1 while pCO2 in bag B ranged from 237 to 395µLL-1 (Fig. 11, grey dots). In both profiles the minimum pCO2 value was observed in the top 5 cm (276 and 237µLL-1, respectively). The maximum pCO2 was observed at 7 cm depth, which corresponds to the direct in situ measurement of brine pCO2 (562 and 599µLL-1, respectively). The bulk ice pCO2 then decreases to reach values of respectively 42 and 350 pLL-1, at the bottom of the ice column. This decrease in bulk ice pCO2 was expected, as sea-water pCO2 in bags A and B was 206 and 300µLL-1, respectively. The very low values in bag A are difficult to understand, but they cannot result from brine loss on sampling, since the technique used (e.g. Reference Tison, Haas, Gowing, Sleewaegen and BernardTison and others, 2002) is designed to prevent it.
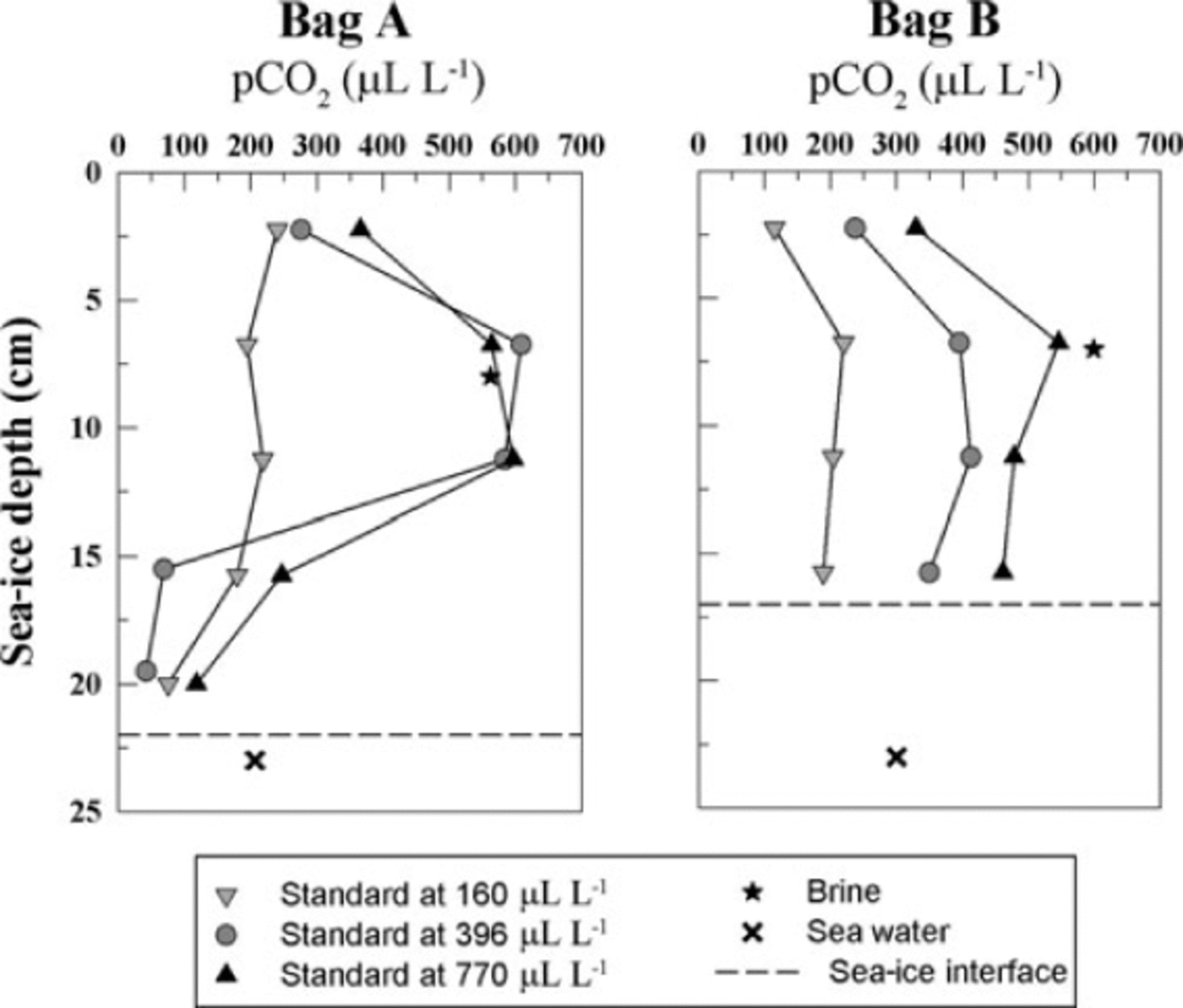
Fig. 11. Profile of pCO2 in bulk sea ice using gas standard of different CO2 concentrations. The sea-ice samples came from Interice IV experiment. The bulk sea-ice concentration was compared to the direct in situ brine pCO2 measurement (black star) and the sea-water concentration (black cross). The dotted line shows the sea-ice interface with the sea water.
Measurements carried out with a 770µLL-1 standard on twin samples collected in the same core at the same level exhibited the same general trend as measurements carried out with a standard of 396 pL L-1. The range of concentration observed was smaller (118-596µLL-1 for bag A and 329546µLL-1 for bag B) and the maximum concentration was in better agreement with the in situ measurement of brine pCO2. Bulk ice pCO2 of bag A was 563µLL-1 while brine pCO2 was 562µLL-1. Bulk ice pCO2 of bag B was 546µLL-1 while brine pCO2 was 599µLL-1. Note that part of the discrepancy between the measurements performed with the two different standards could also result from small-scale spatial variability (e.g. whether the brine channel is included in the sample).
Finally, using a standard gas at 160pLL-1, the general profile of bag A became linear, with a slight decrease of bulk ice pCO2 from 240µLL-1 at the top of the ice to 75µLL-1 in the bottom layer. In bag B, a minimum value of 114µLL-1 is still observed in the top 5 cm. From there down, the profile remains roughly constant (180-220µLL-1). In both cases, the results of bulk ice pCO2 were significantly different from the in situ brine measurement.
This test underlines that attention must be paid to reducing the amount of CO2 exchanged between the standard gas and the brine medium to reach the equilibrium. We must also keep in mind that the pCO2 of the standard should be as close as possible to the bulk ice pCO2.
4. Conclusions
A novel method for obtaining high-resolution pCO2 profiles in bulk sea ice was proposed and tested with respect to its accuracy and reproducibility, both in artificially produced and natural sea-ice samples. The measurements made in artificial standard sea ice demonstrated good reproducibility of the method, compared to a large dataset of brine pCO2 values from Antarctic and Arctic natural environments at temperatures above −9.5°C.
Applying our pCO2 equilibration method in natural sea- ice samples from Barrow provided, to our knowledge, the first-ever published high-resolution pCO2 profiles in sea ice. Both precision and accuracy were demonstrated for the method, provided the ice is permeable at its in situ temperature, i.e. with relative brine volumes above 5%. Below this threshold, repeated measurement of the equilibration standard gas values showed the limitation of the method. Finally more results with different ice types will give more confidence regarding the accuracy and reliability of the method.
The choice of equilibration standard value is a crucial parameter for obtaining reasonably accurate measurements. We suggest that this choice should be based on either an in situ value of brine pCO2 as a first approximation or on the pCO2-temperature relationship proposed in the literature (Geilfus and others, in press). Alternatively, if sufficient material is available, the sample can be measured in triplicate, beginning with an atmospheric standard and refining the choice of the subsequent standards used on the basis of the equilibration pCO2 value from the previous measurements.
This method requires relatively little fieldwork. One core needs to be collected in addition to the temperature and salinity profiles. This method offers the opportunity to work at a high resolution with small ice volumes. It allows us to document small-scale pCO2 gradients and rapid pCO2 variations within the sea-ice cover. It also provides access to a measure of the bulk ice pCO2 at the interface with the atmosphere. Until now, it has been extremely difficult to reconstruct the pCO2 of the upper layer of the sea-ice cover using the sackhole technique. However, this information is crucial for better understanding the CO2 fluxes between sea ice and atmosphere. Nevertheless, as the method is only valid if the ice is permeable, it is currently limited to use in young ice or autumn-spring-summer sea ice.
Acknowledgements
We acknowledge the help of Alan Willam and France Deleu who extensively tested the method. This research was supported by the F.R.S-FNRS (contract 2.4649.07 and 2.4584.09), in which B.D. is a research associate, and the Belgian research program Action de Recherche Concerte ‘Sea Ice Biogeochemistry in a CLIMate change perspective’ (ARC-SIBCLIM) financed by the Belgian French Community under contract No. ARC-02/7-287. N.-X.G. received a PhD grant from the Fonds pour la Formation a la Recherche dans l’Industrie et l’Agriculture. Work carried out in the frame of the Interice IV experiment was supported by the European Community’s Sixth Framework Program through the grant to the budget of the Integrated Infrastructure Initiative HYDRALAB III, contract No. 022441 (RII3). We thank the Hamburg Ship Model Basin (HSVA), especially the ice tank crew, for hospitality, technical and scientific support and professional execution of the test program in the Research Infrastructure ARCTECLAB. We are also indebted to Hajo Eicken and the rest of the Geophysical Institute of the University of Alaska Fairbanks, the North Slope Borough and the Barrow Arctic Science Consortium for logistical support during field survey work carried out in Barrow. We thank two anonymous reviewers for constructive comments that helped to improve the manuscript. This is MARE contribution No. 221 (MARE is the interfaculty Center for Marine Research, University of Liège).












