



Introduction
Obesity is a central driver of cardiometabolic syndrome, a cluster of chronic disorders that includes type 2 diabetes mellitus (T2DM), dyslipidemia, and cardiovascular diseases (CVD) Reference Skinner, Perrin, Moss and Skelton1 ; it is a silent pandemic that produces immense health and economic burdens Reference Bluher2 . In the US alone, the WHO estimates that by 2030, 50% of people will be obese and 25% morbidly obese Reference Ataey, Jafarvand, Adham and Moradi-Asl3 , making >50% of the US population at risk of developing obesity-associated chronic diseases. Addressing the obesity pandemic is of paramount importance, albeit a tremendous challenge as the disease exists in diverse forms and its primary pathogenic factors may include genetic abnormalities, adverse lifestyle factors, and early-life exposure to adverse conditions Reference Bluher2 .
In 2018, more than 65% of US women of childbearing age were either overweight or obese Reference Catalano and Shankar4 , and it is now believed that obesity during pregnancy is a major driving force behind the obesity pandemic Reference Gambineri, Conforti and Di Nisio5 . Intrauterine exposure to maternal obesity is linked to development of obesity and associated chronic diseases later in life – a process called developmental programming 6. This induces a vicious cycle of obesity: daughters of obese mothers have increased likelihood of developing obesity themselves, which, in turn, predisposes their future children to obesity Reference King, Dakin and Liu7 . However, while the phenomenon of developmental programming is well-described, the precise mechanisms, by which it is mediated, remain unclear. This knowledge gap poses a major obstacle to the development of effective strategies for addressing the generational perpetuation of obesity programming.
Dipeptidyl peptidase IV (DPPIV), also known as CD26, is a serine protease that has crucial roles in the regulation of metabolism, appetite, food intake, energy expenditure, and body composition Reference Varin, Mulvihill and Beaudry8 . DPPIV is produced by hepatocytes, adipocytes, intestinal K cells, placental cytotrophoblasts, and endothelial cells, among others, and exists in both soluble (s-DPPIV) and plasma membrane-bound (m-DPPIV) isoforms Reference Klemann, Wagner, Stephan and von Hörsten9 . These two isoforms have distinct functions: m-DPPIV regulates intercellular communication by acting as a receptor, while s-DPPIV facilitates intercellular and inter-organ crosstalk by cleaving protein substrates in bodily fluids such as plasma and/or binding to DPPIV receptors on cell membranes Reference Klemann, Wagner, Stephan and von Hörsten9 . In the context of obesity, DPPIV has been shown to exacerbate fat retention and metabolic abnormalities Reference Lamers, Famulla and Wronkowitz10–Reference Sarkar, Nargis, Tantia, Ghosh and Chakrabarti13 . Evidence from epidemiological studies revealed that plasma DPPIV activity is increased in obese children Reference Iwabuchi, Kamoda and Saito14 , in obese adults Reference Sanz, Larrinaga and Fernandez-Atucha12 , and in patients with T2DM Reference Lee, Jang, Song, Kim and Kim11–Reference Sarkar, Nargis, Tantia, Ghosh and Chakrabarti13,Reference Williams, Vieira De Ribeiro and Prakoso15 , hypertension Reference Balfour, Rodriguez and Ferdinand16 , and renal diseases Reference Lovshin and Zinman17,Reference Kanasaki18 . Furthermore, emerging evidence suggests that elevated plasma DPPIV activity in childhood is a marker of increased risk for later-life obesity Reference Sanz, Larrinaga and Fernandez-Atucha12,Reference Sarkar, Nargis, Tantia, Ghosh and Chakrabarti13,Reference Koh19 . In mice, greater plasma DPPIV activity is observed in models of genetic obesity (ob/ob) and diet-induced obesity (DIO) Reference Schurmann, Linke and Engelmann-Pilger20 ; meanwhile, mice that lack one or both Dpp4 alleles in adipocytes and/or hepatocytes exhibit reduced plasma DPPIV activity and are protected from DIO Reference Takeda, Sawazaki and Takahashi21 . DPPIV has also been extensively studied in the context of T2DM and insulin signaling, and DPPIV inhibitors such as sitagliptin are effective, United States Food and Drug Administration (FDA)-approved second-line treatments for T2DM Reference Yoshikawa, Tsuchiya and Kido22–Reference Tang, Huang, Liu, Shankar, Ganz and Rajpathak27 . In line with the hypothesis that DPPIV is an active player in obesity progression, results from clinical trials indicate that DPPIV inhibitors can improve weight control Reference Lipscombe28–Reference Waters, Topp, Siler and Alexander34 , systemic metabolism Reference Yoshikawa, Tsuchiya and Kido22–Reference Tang, Huang, Liu, Shankar, Ganz and Rajpathak27 , and inflammatory responses Reference Ferreira, Teixeira-de-Lemos and Pinto35–Reference Prakash, Rai, Kosuru, Tiwari and Singh43 – all critical processes that become progressively dysregulated in the context of obesity. Nonetheless, despite extensive research efforts, the precise role of DPPIV in obesity progression remains to be determined.
In this respect, whether DPPIV has a role in developmental programming is an entirely novel direction of inquiry. Here, we determined the effect of maternal obesity on maternal and offspring plasma DPPIV activity in humans and non-human primates, and further tested the hypothesis that DPPIV plays a critical role in the progression of obesity and glucose intolerance in male offspring of obese mothers, and that inhibition of DPPIV will have potential as preventive therapeutics against obesity-related developmental programming. Our findings indicate that DPPIV activity is increased in obese mothers, and also in their offspring in an offspring sex- and age-dependent manner. Using a mouse model of high-fat diet (HFD)-induced maternal obesity, we show that administration of the DPPIV inhibitor sitagliptin to HFD-fed mothers during the immediate perinatal period delays progression of obesity and metabolic disorders in their male offspring, while female offspring remain unaffected. Importantly, our findings highlight a critical role for DPPIV in developmental programming of maternal obesity and provide supporting evidence that DPPIV inhibitors merit further examination as potential therapeutics to address developmental programming and the progression of obesity and metabolic disorders in offspring of obese mothers.
Results
Maternal obesity dysregulates plasma DPPIV activity in humans and non-human primates
To determine if maternal obesity has an effect on plasma DPPIV activity in humans, we collected maternal and cord blood plasma from normal-weight (Hum-Mat-NW, body mass index [BMI]<25, n = 25) and obese (Hum-Mat-Ob, BMI > 30, n = 13) mothers upon C-section at term. The clinical characteristics of the study participants are presented in Table 1. Indications for Caesarean section included only elective repeat or breech presentation, and no differences in indications were observed between normal-weight and obese groups. By experimental design, the groups differed significantly in terms of BMI. No differences were observed in maternal and gestational age, birth weight, or gestational weight gain.
Table 1. Clinical information of human donors of maternal and cord blood samples. Data presented as mean (range). Sample size is shown. *, p < 0.05

Using an amino-methylcoumarin fluorogenic assay Reference Lorey, Faust, Buhling, Ansorge and Neubert44 , we found that relative to normal-weight mothers, plasma DPPIV activity was significantly increased in obese mothers carrying male fetuses but decreased in mothers with female fetuses (Fig. 1A). Cord plasma DPPIV activity was likewise increased in male babies born to obese mothers but showed no difference in females (Fig. 1B). Next, we asked whether this activation of DPPIV in offspring persists beyond birth. Due to a lack of human samples, we addressed this question using two well-described non-human primate models of maternal diet-induced developmental programming: baboons (Bab, Papio hamadryas) and Japanese macaques (JM). Both of these model primate species have been extensively studied in the setting of maternal obesity due to high genetic and physiological similarities with humans Reference Booz, Massoud, Altara and Zouein45,Reference Disotell and Tosi46 . Phenotype details for the baboon Reference Schlabritz-Loutsevitch, Lopez-Alvarenga and Comuzzie47–Reference Huber, Jenkins, Li and Nathanielsz51 and Japanese macaque Reference True, Arik, Lindsley, Kirigiti, Sullivan and Kievit52–Reference Campodonico-Burnett, Hetrick and Wesolowski56 models of maternal HFD feeding vs. maternal regular diet (RD) are extensively described in the cited reports. In both models, the offspring of HFD-fed mothers (Off-HFD) in relation to offspring of a RD-fed mothers (Off-RD), exhibit programmed diseases whose development bears a striking resemblance to those occurring in human offspring of obese mothers. In six-month-old (pre-weaning) baboons both male and female offspring of HFD-fed mothers (Bab-Off-HFD) exhibited increased plasma DPPIV activity relative to offspring of RD-fed mothers (Bab-Off-RD) (Fig. 1C). Similarly, in three-year-old (pre-pubescent) JM, we found increased plasma DPPIV activity in both male and female offspring of HFD-fed mothers (JM-Off-HFD) relative to offspring of RD-fed mothers (JM-Off-RD) (Fig. 1D).
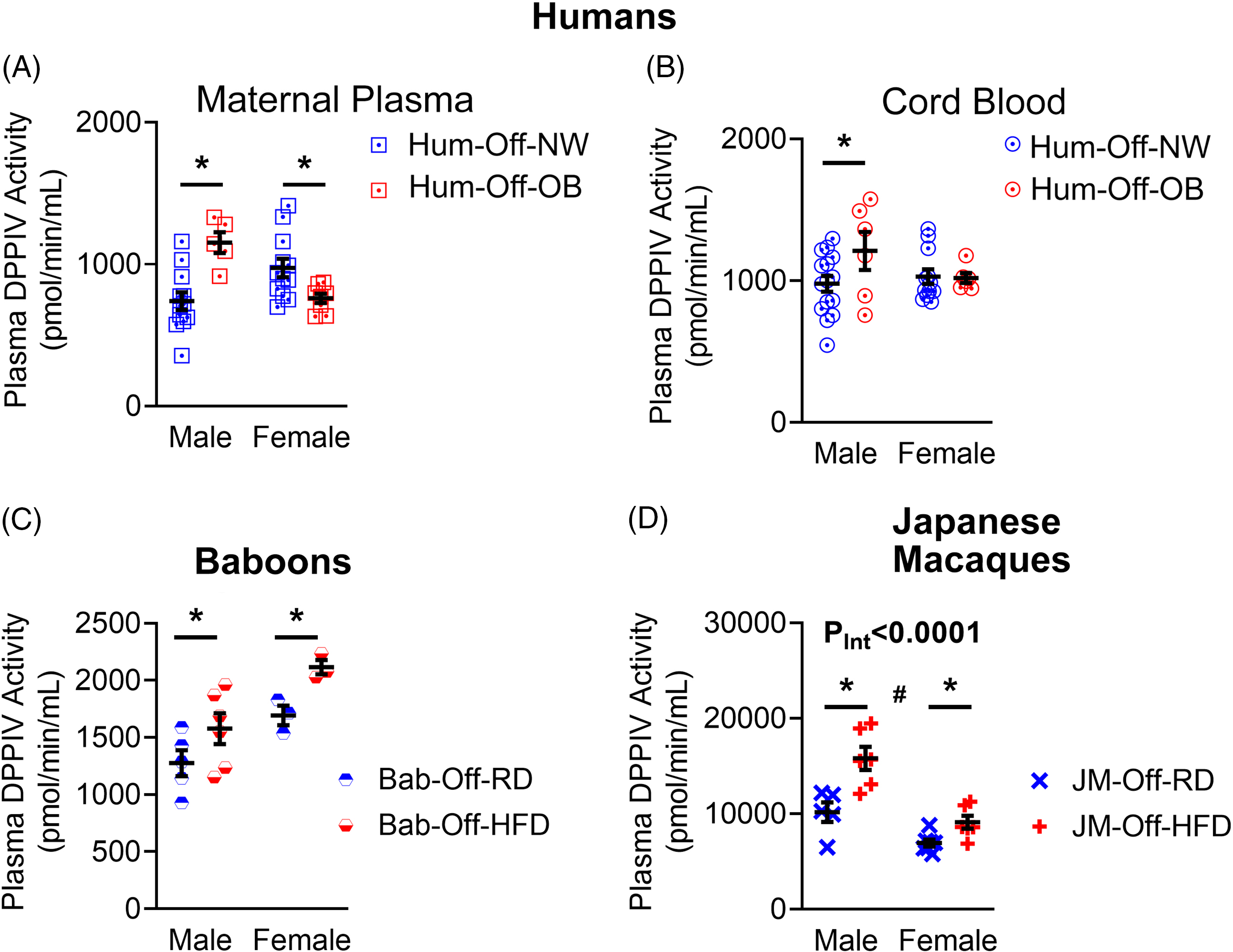
Fig. 1. Maternal obesity dysregulates plasma activity of dipeptidyl peptidase IV in humans and non-human primates. DPPIV activity was measured in maternal plasma samples (A) from normal-weight (Hum-Mat-NW) and obese (Hum-Mat-Ob) mothers at term and prior to delivery by C-section, and in cord plasma (B) from male and female fetuses born to normal-weight (Hum-Off-NW) and obese mothers (Hum-Off-Ob). Sample size is shown in Table 1. *, p < 0.05 NW vs. OB women. C-D, Plasma DPPIV activity was measured in six-month-old male and female baboon offspring (C) of regular diet (RD)-fed mothers (Bab-Off-RD, n = 6 males and 3 females) and high-fat diet (HFD)-fed mothers (Bab-Off-HFD, n = 6 males and 3 females), and in 36-month-old male and female Japanese macaque offspring (D) of RD-fed (JM-Off-RD, n = 5 males and 7 females) and HFD-fed (JM-Off-HFD, n = 6 males and 6 females) mothers. *, p < 0.05 offspring of RD-fed mothers vs. offspring of HFD-fed mothers.
Mouse model of maternal high fat DIO
To determine if activation of DPPIV contributes to developmental programming, we developed a mouse model of maternal HFD-induced obesity. Specifically, we fed female wild-type Friend virus B (FVB/NJ) mice either a RD (13% kcal from fat) or a HFD (45% kcal from fat) starting from six weeks of age and continuing throughout the study (Fig. 2A). Composition of diets is shown in Table S1. After eight weeks of dietary intervention (i.e. at 14 weeks of age) and prior to breeding, HFD-fed females (Mat-HFD) exhibited no change in body weight (Fig. 2B) relative to RD-fed females (Mat-RD) but had increased body fat composition (Fig. 2C) as visualized using microcomputed (Fig. 2C) measured and quantified using echo magnetic resonance (EchoMRI, Fig. 2D). To confirm that feeding a HFD does not lead to pre-gestational diabetes, we assessed glycemic control using both an oral glucose tolerance test (OGTT) (Fig. 2E and F) and an insulin tolerance test (Fig. 2G and H). No significant differences were observed between Mat-RD and Mat-HFD in their responses to either test.
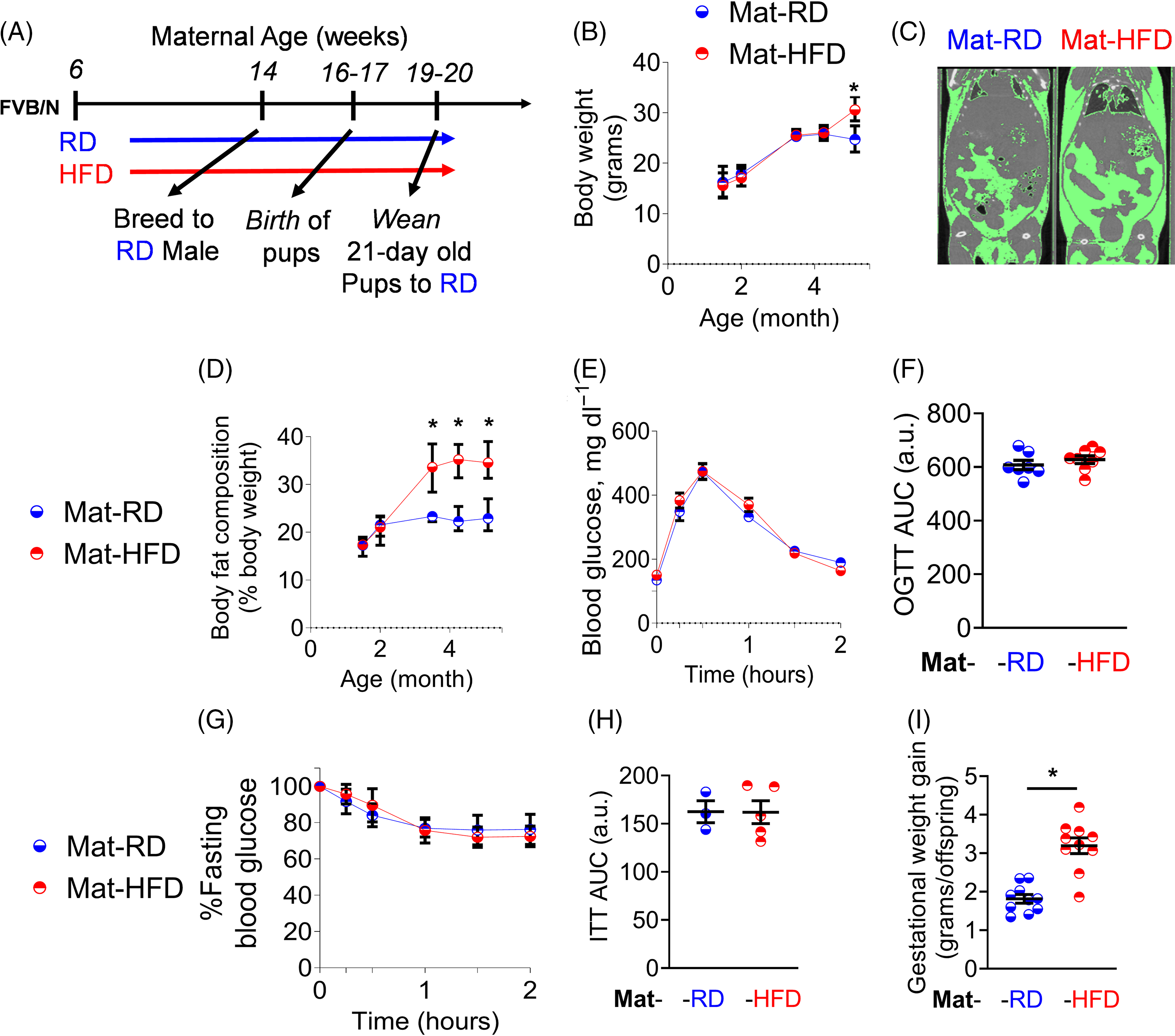
Fig. 2. HFD feeding of female mice leads to increased maternal adiposity without metabolic dysfunction. Study design (A). Panels illustrate body weights (B), representative microcomputed tomography radiographs (C), body fat composition (D), OGTT glycemic excursion curves (E), OGTT AUC values for E (F), ITT glycemic excursion curves (G), ITT AUC (H), and gestational weight gain (I) of Mat-RD (n = 7) and Mat-HFD (n = 8). *, p < 0.05 HFD-fed females vs. RD-fed females.
After eight weeks of dietary intervention, 14-week-old RD- and HFD-fed females were bred to RD-fed males. During pregnancy, the Mat-HFD group gained more weight than did Mat-RD females (Fig. 2I). At birth, pups born to HFD-fed mothers (Off-HFD) exhibited a 16% reduction in birthweight compared with pups born to RD-fed mothers (Off-RD) (Fig. 3A, p < 0.001). Pups were weaned at three weeks and thereafter were fed only the RD. As of weaning and into pre-pubescence (two months of age), no significant differences in body weight were evident in either male or female Off-HFD relative to Off-RD (Fig. 3B-C); however, at adulthood (i.e. from 4 through 11 months of age), both male and female Off-HFD were continuously heavier than their Off-RD counterparts (Fig. 3B-C).

Fig. 3. In mice, maternal HFD and obesity leads to programming of obesity and metabolic dysfunction in male offspring. Data were collected from offspring of mothers that underwent the experimental protocol shown in Fig. 2A. Average birthweight of Off-RD and Off-HFD pups in each litter (A). N = 10/group of maternal diet. Body weights of male (B) and female (C) Off-RD/-HFD at three weeks old and at two, four and 11 months old. Representative microcomputed tomography radiographs at two months of age (D) and EchoMRI body fat mass quantification at four months of age (E). OGTT glycemic excursion curves of three-week-old male (F) and female (G) Off-RD/HFD, and OGTT AUC values (H). OGTT glycemic excursion curves of two-month-old male (I) and female (J) Off-RD/-HFD, and AUC values (K). ITT glycemic excursion curves of two-month-old male (L) and female (M) Off-RD/-HFD, and AUC values (N). OGTT glycemic excursion curves of 11-month-old male (O) and female (P) Off-RD/HFD, and AUC values (Q). N = 10/group/sex. *, p < 0.05 offspring of HFD-fed mothers vs. offspring of RD-fed mothers. #, p < 0.05 males vs. females within the same group of maternal diet.
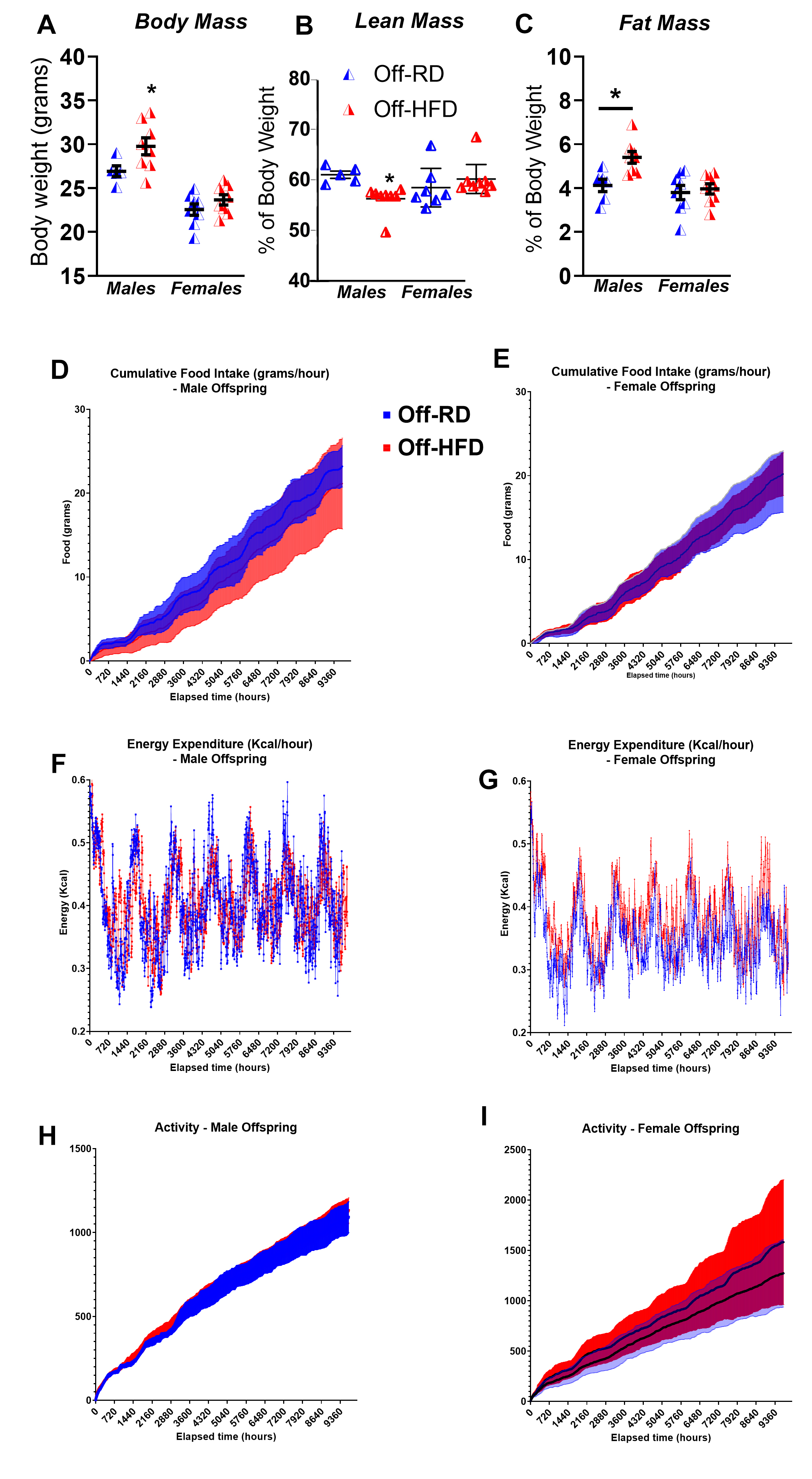
At two months of age, we visualized offspring body composition using microcomputed tomography, which revealed increased fat mass in Off-HFD males and to a lesser degree in Off-HFD females (Fig. 3D). At four months of age, we measured body composition again using EchoMRI and found total fat mass to be significantly increased and lean mass decreased (p < 0.05 for both) in Off-HFD males with no changes in females (Supp. Fig. 1A-C). As adiposity in the Off-HFD could be attributed to changes in ‘lifestyle choices’ such as food intake, energy expenditure, or overall physical activity, we performed metabolic and behavioral phenotyping in four-month-old Off-RD and Off-HFD mice using indirect calorimetry (Promethion systems) (Supp. Fig. 1). We observed no significant differences between the groups in terms of food intake (Supp. Fig. 1D-E), energy expenditure (Supp. Fig. 1F-G), or overall activity (Supp. Fig. 1H-I), suggesting that the increased adiposity in Off-HFD was induced by exposure to maternal HFD, and is not a direct effect of lifestyle factors such as food consumption and/or physical activity.
Since children born to obese mothers are prone to developing metabolic dysregulations Reference Herring and Oken57 , we measured glucose tolerance and insulin sensitivity in male and female Off-RD and Off-HFD mice across a range of ages. At three weeks of age, glucose tolerance appeared comparable between groups for males and females alike (Fig. 3F-H). At two months of age, however, male Off-HFD exhibited a 44% decrease in glucose tolerance (Fig. 3I,K) and a 34% decrease in insulin sensitivity (Fig. 3L,N) relative to their Off-RD counterparts (p < 0.05), whereas no significant changes were evident in females (Fig. 3J,K,M,N). At 11 months of age, male Off-HFD likewise exhibited 40% decreased glucose tolerance (Fig. 3O,Q) along with OGTT glycemic excursion curves similar to mice having type 2 diabetes Reference Winzell and Ahrén58 , while females continued to demonstrate no significant difference in either measure (Fig. 3P-Q) suggesting that Off-HFD male offspring appearing to be more metabolically compromised than female offspring through at least 11 months of age.
A maternal HFD in mice leads to activation of offspring plasma dipeptidyl peptidase DPPIV
Having established our mouse model of maternal obesity, we next examined the effect of maternal HFD on plasma activity of DPPIV. After eight weeks of dietary intervention and prior to breeding, HFD-fed females showed a tendency to increased plasma DPPIV activity but did not achieve statistical significance (p = 0.1, Fig. 4A). In three-week-old offspring, we found that plasma DPPIV activity was significantly increased in Off-HFD males relative to Off-RD males; these results are similar to observations in human babies, and suggest that DPPIV activation precedes metabolic dysregulations in these mice (Fig. 4B). Significantly reduced DPPIV activity was observed in three-week-old Off-HFD females (p < 0.05, Fig. 4B), but at 11 months of age, both male and female Off-HFD showed significant increases in DPPIV plasma activity (p = 0.007 and p = 0.05 respectively, Fig. 4C). These results indicate that, consistent with human and non-human primate data, maternal HFD-induced adiposity in mice dysregulates plasma DPPIV activity in a manner dependent on offspring sex and age. Hence, we reasoned that DPPIV might play a role in the developmental programming of obesity and metabolic diseases, at least in the maternal HFD context.

Fig. 4. In mice, maternal HFD dysregulates offspring plasma DPPIV activity in a sex- and age-dependent fashion, and acute DPPIV inhibition improves glucose tolerance in male Off-HFD. Plasma DPPIV activity of 14-week-old RD- and HFD-fed female FVB/n mice (A), and of three-week-old (B) and 11-month-old (C) Off-RD (n = 5-8/sex) and Off-HFD (n = 6-11/sex). Data in D-I were collected from two- and 11-month-old male and female Off-RD (n = 4-8/sex) and Off-HFD (n = 5/sex). Data for untreated Off-RD/-HFD groups were previously presented in Figure 2. OGTTs were first conducted on untreated mice (Off-RD/-HFD), then after a two-day rest were repeated on the same mice after one-hour pretreatment with sitagliptin (i.p. 30 mg/kg) (Off-RD/-HFD + Ac-Sita). OGTT glycemic excursion curves and respective AUC quantifications for two-month-old males (D, E), 11-month old males (F, G) and 11-month-old females (H, I). *, p < 0.05 offspring of HFD-fed mothers vs. offspring of RD-fed mothers. #, p < 0.05 males vs. females within the same group of maternal diet.
Administration of the DPPIV inhibitor sitagliptin to male offspring of HFD-fed mothers ameliorates progression of obesity and improves glycemic control
To determine if DPPIV is involved in the metabolic abnormalities observed in Off-HFD mice, we treated the same cohorts of 2- and 11-month-old Off-RD and Off-HFD mice that were assayed for glucose control, with sitagliptin, a DPPIV inhibitor, (acute treatment, Ac-Sita, i.p. 0.3 mg/kg), then one hour later repeated the glucose tolerance test (Fig. 3). Sitagliptin administration noticeably improved glucose tolerance in both Off-RD and Off-HFD males, by 39 and 49% respectively at two months of age (Fig. 4D and E) and by 3.9% and 11.2% respectively at 11 months of age (Fig. 4F and G). No significant effects of Ac-Sita treatment were observed in females (Fig. 4H and I).
Next, we asked whether chronic administration of sitagliptin from a young age would similarly improve glycemic control and furthermore prevent the progression of obesity and metabolic abnormalities in Off-HFD mice. We treated male and female Off-RD and Off-HFD with sitagliptin (Sita, 0.3 mg/kg) or vehicle (Veh, DMSO) via drinking water, beginning at three weeks of age and extending into adulthood. No differences in the amount of water consumed each day were observed between sexes or between experimental groups.
At four months old, the Off-HFD vehicle-treated (Off-HFD + Veh) males exhibited a 7% increase in body weight (Fig. 5A) and a 56% increase in body fat percentage (measured as the percentage of body weight accounted for by inguinal adipose tissue) (Fig. 5C) compared with their Off-RD counterparts (Off-RD + Veh). However, sitagliptin-treated Off-HFD males showed a 50% reduction in body weight (p < 0.05, Fig. 5A) and 44% reduction in adiposity vs. Off-HFD + Veh (p < 0.05, Fig. 5C). Meanwhile, no differences in body weight or body composition were observed in four-month-old Off-HFD females, whether treated with sitagliptin or vehicle (Fig. 5B). Surprisingly, in Off-RD females, sitagliptin treatment reduced adiposity by 46% (p = 0.04, Fig. 5D) vs. vehicle-treated mice.

Fig. 5. DPPIV inhibitor treatment of male Off-HFD mice downregulates weight gain, glucose intolerance, and insulin resistance. Data were collected from Off-RD (n = 4-5/sex) and Off-HFD (n = 5/sex) that were treated with sitagliptin (+Sita) at a dosage of 30-45 mg/kg/day or its vehicle (+Veh, DMSO) in drinking water from weaning (three-weeks-old) until endpoint. Body weight trends of male (A) and female (B) offspring from all groups. OGTT glycemic excursion curves (E, F) and respective AUC quantifications (G, H) of two-month-old male (E, G) and female (F, H) offspring from all groups. ITT glycemic excursion curves (I-J) and respective AUC quantifications (K, L) of two-month-old male (I, K) and female (J, L) offspring from all groups. *, p < 0.05 offspring of HFD-fed mothers vs. offspring of RD-fed mothers. &, p < 0.05 sitagliptin-treated mice vs. vehicle-treated mice.
To determine the effects of chronic sitagliptin treatment on glycemic control, we measured glucose tolerance and insulin sensitivity at two months of age and compared the results with previously collected data (Fig. 3). Sitagliptin-treated Off-HFD males showed significant improvement, with the area under the curve for the glucose tolerance test being decreased by 10.4% (p = 0.03, Fig. 5E,G) and that for the insulin sensitivity test by 11% (p = 0.05, Fig. 5I,K) relative to vehicle-treated Off-HFD males. In females, neither glucose tolerance (Fig. 5F,H) nor insulin sensitivity (Fig. 5J,L) were affected by maternal obesity, and likewise no changes were observed with sitagliptin treatment.
DPPIV inhibition during pregnancy and lactation in HFD-fed mothers delays progression of obesity and metabolic dysfunction in male offspring
In light of the results obtained from long-term sitagliptin treatment in offspring, we hypothesized that maternal inhibition of DPPIV would impede the progression of obesity and metabolic disorders in offspring. To address this hypothesis, we conducted a pre-clinical study, in which HFD- or RD-fed female mice were administered with either sitagliptin (Sita, 0.3 mg/kg) or vehicle (Veh, DMSO) starting one week prior to breeding and continuing throughout pregnancy and lactation (study design is shown in Fig. 6A). This treatment regimen is henceforth referred to as “Mat-Sita.”

Fig. 6. DPPIV inhibitor treatment of HFD-fed female mice does not affect gross morphology and glycemic control, but decreases gestational weight gain. Study design (A), in which RD- and HFD-fed mice were treated with either sitagliptin (+Sita) at a dosage of 30-45 mg/kg/day or its vehicle (+Veh) (DMSO) in drinking water. Body weights (B), body fat composition (C), gestational weight gain (C), OGTT glycemic excursion curves (D), and OGTT AUC values (E) of mothers from all groups. N = 4-7/group. *, p < 0.05 offspring of HFD-fed mothers vs. offspring of RD-fed mothers. #, p < 0.05 offspring of sitagliptin-treated mothers vs. offspring of vehicle-treated mothers.
In RD and HFD-fed mothers, Sita did not affect body weight (Fig. 6B) or glucose tolerance prior to pregnancy (Fig. 6D and E) when compared with respective Veh-treated controls. Meanwhile, gestational weight gain was decreased by Sita in HFD-fed mothers (Mat-HFD + Sita) vs. Veh-treated HFD-fed mothers (Mat-HFD + Veh), with no effect observed in RD-fed mothers (Fig. 6C). Sita treatment likewise had no effect on birthweight in either Off-RD or Off-HFD mice (Fig. 7A), and reduced body weights at weaning in males (p < 0.05, Fig. 7B) with no changes in females (Fig. 7C) pups. However, where two-month-old male offspring of the Mat-HFD + Veh group showed a 25% increase in body weight vs. Mat-RD + Veh (p < 0.05), that change was ameliorated in the male offspring of Mat-HFD + Sita mice (p < 0.05, Fig. 7D). No changes were observed in females (Fig. 7C,E). Interestingly, at ten months of age, both male and female offspring of the Mat-HFD + Veh group demonstrated significantly higher body weights (Fig. 7F and G) and body fat composition (Fig. 7H and I), that were reversed in the offspring of Mat-HFD + Sita mothers. No changes in lean mass were detected under either treatment.

Fig. 7. In mice, administration of DPPIV inhibitor to HFD-fed mothers delays progression of obesity in male offspring. Data were collected from male and female offspring of mothers that underwent the experimental protocol illustrated in Fig. 6A. Average birthweights of all pups in litters from all groups (A). Body weights of three-week-old males (B) and females (C), two-month-old males (D) and females (E), and ten-month-old males (F) and females (G) from all offspring groups. Body fat composition of ten-month-old males (H) and females (I) from all offspring groups. Plasma DPPIV activity of three-week-old males (J) and females (K) from all offspring groups. N = 5-15/group/sex. #, p < 0.05 offspring of sitagliptin-treated mothers vs. offspring of vehicle-treated mothers.
To determine if maternal administration of sitagliptin affects DPPIV activity in young offspring, we assessed plasma DPPIV activity at weaning. Consistent with the results illustrated in Fig. 5, we found maternal HFD to increase plasma DPPIV activity in male offspring and decrease it in female offspring relative to sex-matched offspring of RD-fed mothers (p = 0.01, Fig. 7J). Maternal Sita administration decreased plasma DPPIV activity in 3-week-old male pups born to HFD-fed mothers but not in those born to RD-fed mothers. Surprisingly, for female offspring, maternal Sita treatment decreased plasma DPPIV activity in pups born to RD-fed mothers and increased it in pups born to HFD-fed mothers (p = 0.003) (Fig. 7K).
To investigate if maternal sitagliptin modifies offspring glycemic control, we measured offspring glucose tolerance and insulin sensitivity at weaning (three weeks of age), two months, and ten months of age. At weaning, maternal HFD did not affect glucose tolerance in either Veh- or Sita-treated groups, and Mat-Sita improved glucose tolerance by 24% in male Off-RD mice (p = 0.01) but not in any other group (Fig. 8A-D). In Veh-treated groups at two months of age, male Off-HFD mice exhibited significant impairment in both glucose tolerance (Fig. 8E,G) and insulin sensitivity (Fig. 8M,O) vs. their Off-RD counterparts, while female offspring exhibited no changes in either glucose tolerance (Fig. 8F,H) or insulin sensitivity (Fig. 8N,P). Importantly, Mat-Sita improved glucose tolerance in both male and female Off-HFD mice, by 21% (Fig. 8E,G) and 28% (Fig. 8F,H) respectively, while insulin sensitivity was unaffected (Fig. 8M-P). At ten months of age, both male (Fig. 8I,K) and female (Fig. 8J,L) offspring of Veh-treated HFD-fed mothers exhibited increased glucose intolerance (28 and 17% respectively), but Mat-Sita treatment produced no significant effect (Fig. 8I-L).

Fig. 8. In mice, DPPIV inhibitor treatment of HFD-fed mothers delays progression of metabolic abnormalities in male offspring. Data were collected from male and female offspring of mothers that underwent the experimental protocol illustrated in Fig. 6A. OGTT glycemic excursion curves and respective AUC quantifications of three-week-old males (A, C) and females (B, D), two-month-old males (E, G) and females (F, H), and ten-month-old males (I, K) and females (J, L) from all offspring groups. ITT glycemic excursion curves and respective AUC quantifications of two-month-old males (M, O) and females (N, P) from all offspring groups. N = 5-15/group/sex. #, p < 0.05 offspring of sitagliptin-treated mothers vs. offspring of vehicle-treated mothers.
Discussion
Exposure to an adverse intrauterine environment such as maternal obesity is now recognized as a major driving force behind the rapidly rising incidence of obesity and associated chronic diseases. However, the underlying mechanisms by which maternal obesity programs obesity in offspring yet remain unclear, and there are consequently no effective strategies for addressing the passage of obesity from obese mothers to their children. A major goal in treating such detrimental developmental programming is to delay and/or reverse the progression of programmed diseases. Here, we found that plasma DPPIV activity is increased in male offspring of obese mothers. Using two well-established non-human primate models of maternal obesity, we further revealed that such dysregulation can persist into adult life for offspring born to obese mothers. Hence, we reasoned that DPPIV likely plays an active role in developmental programming mediated by maternal obesity. To investigate the effects of pharmacologic DPPIV inhibition on the progression of programmed offspring obesity, we performed a pre-clinical study in a mouse model of maternal high fat DIO for which we used the FDA-approved anti-diabetic drug sitagliptin to inhibit DPPIV activity in vivo. We demonstrated that both long-term DPPIV inhibition in offspring starting from a young age and short-term maternal inhibition of DPPIV during gestation and lactation blunt the progression of obesity and metabolic disorders in male offspring of HFD-fed mothers. Thus, our findings highlight dysregulation of DPPIV as being at least partly responsible for the sex-dependent programming of obesity and metabolic disorders in offspring of obese mothers. Furthermore, we suggest that DPPIV inhibitor therapy could be a putative strategy for halting and/or inhibiting this process.
Our finding that maternal obesity leads to dysregulation of plasma DPPIV activity in both mother and offspring is not surprising. DPPIV is strongly linked to obesity Reference Sanz, Larrinaga and Fernandez-Atucha12 ; prior studies have reported increased s-DPPIV in obese adults Reference Lee, Jang, Song, Kim and Kim11 , in lean children that subsequently develop obesity Reference Iwabuchi, Kamoda and Saito14 , and in mice with DIO Reference Pospisilik, Ehses, Doty, McIntosh, Demuth and Pederson59,Reference Conarello, Li and Ronan60 . Both obesity and/or hypercaloric diets have been shown to increase levels of plasma DPPIV by upregulating production of s-DPPIV and/or by accelerating shedding of m-DPPIV Reference Williams, Vieira De Ribeiro and Prakoso15,Reference Mulvihill, Varin and Gladanac61 from DPPIV-expressing cells. Moreover, children born to obese mothers often exhibit abnormalities in regulation of blood glucose, blood lipids and body fat mass Reference Herring and Oken57 – all of which are regulated by DPPIV. In the present study, one of our novel findings is that early-life dysregulation in plasma DPPIV activity precedes metabolic abnormalities in male Off-HFD mice, suggesting that DPPIV could be a culprit in the progression of programmed diseases stemming from maternal obesity. Furthermore, our observation that DPPIV is dysregulated in both obese mothers carrying male fetuses and their newborn male offspring suggests that DPPIV could be involved in developmental programming in males even prior to birth.
Emerging evidence suggests that DPPIV activation in the setting of obesity is driven by excessive cellular workload or cellular stress and manifests within tissues as increased expression of endoplasmic reticulum (ER)-stress markers, accumulation of reactive oxygen species (ROS), and production of pro-inflammatory cytokines Reference Ghorpade, Ozcan and Zheng63 . DPPIV is constitutively expressed in hepatocytes, endothelial cells, adipocytes immune cells, intestinal K cells, and placental cytotrophoblasts, among others; more than 90% of plasma s-DPPIV is derived from these cell types. Importantly, a growing body of evidence suggests that the cellular origin of s-DPPIV determines its activity and therefore its physiological effect. For instance, Mulvihill et al. Reference Maloyan, Muralimanoharan and Huffman49 demonstrated that while enterocyte DPPIV plays no role in glucose homeostasis, hematopoietic DPPIV can cleave the incretin hormone gastric inhibitory peptide (GIP) but not glucagon-like peptide 1 (GLP-1), and endothelial s-DPPIV can degrade both hormones in insulin-resistant states. In a 2018 study using a mouse model of DIO, Ghorpade et al. demonstrated that hepatocyte-secreted s-DPPIV and factor-Xa cooperatively activate inflammation in adipose tissue macrophages, leading to obesity and insulin resistance Reference Ghorpade, Ozcan and Zheng63 . It is yet unclear what exactly triggers the observed dysregulation of plasma DPPIV activity in obese mothers, from which cell/tissue the dysregulated enzyme is derived, and how the sex of the fetus determines the direction in which the dysregulation of maternal DPPIV will occur. Work to answer these important questions is presently underway in our laboratory.
In the meantime, we posit that maternal DPPIV may contribute to developmental programming in three ways. First, dysregulation of maternal m- and s-DPPIV can disrupt critical pregnancy-induced physiological states – the most important of which is maternal insulin resistance. Inter-organ crosstalk mediated by s-DPPIV regulates insulin production by degrading incretins Reference Mulvihill and Drucker62 and insulin sensitivity Reference Mulvihill, Varin and Gladanac61,Reference Ghorpade, Ozcan and Zheng63 ; as such, abnormalities in maternal DPPIV may directly alter control of maternal physiological states, which in turn indirectly affects fetal development. While we observed obese and normal-weight human mothers to have no differences in insulin sensitivity (measured by HOMA-IR) Reference Bucher, Montaniel, Myatt, Weintraub, Tavori and Maloyan64 , our approach is limited in that maternal plasma was only collected at term and therefore our data are not informative regarding maternal insulin sensitivity throughout gestation. Second, dysregulation of maternal s-DPPIV could impair fetal-maternal crosstalk through the placenta. Although the role of DPPIV in such crosstalk has not been studied, it is likely that maternal plasma s-DPPIV affects cellular processes in the placenta. It has been shown that maternal s-DPPIV can cross the placenta (Januvia [package insert]. Kenilworth, NJ: Merck Pharmaceuticals, Inc. 2010), and that DPPIV receptors such as caveolin-1 are expressed on fetal stem cells Reference Baker and Tuan65 . Accordingly, we posit that as maternal blood perfuses placental capillaries, maternal plasma s-DPPIV will bind to caveolin-1 on the membrane of placental cytotrophoblasts Reference Naing, Hamilton, van Zuylen, Scott and Rawlinson66 , thereby inducing intracellular signaling cascades; in effect, this represents a communication axis between s-DPPIV-producing maternal cells and placental cytotrophoblasts. Ongoing studies in our laboratory focus on addressing the role of DPPIV in placental function, and our preliminary results show reduced mitochondrial respiration in cytotrophoblasts treated with DPPIV (Montaniel, Bucher & Maloyan, manuscript in preparation). As such, abnormalities in placental function represent a second possible consequence of dysregulated maternal s-DPPIV; such abnormalities have been previously linked to developmental programming67,68. Third, dysregulation of maternal s-DPPIV could permanently alter the epigenomic landscape of offspring stem cells, including mesenchymal, adipocyte, and hematopoietic stem cells, thereby altering the fate and phenotype of their future daughter cells, ultimately affecting essentially all cells in critical metabolic organs such as the liver and adipose tissues Reference Boyle, Patinkin and Shapiro69,Reference Baker, Patinkin, Shapiro, De La Houssaye, Woontner and Boyle70 . Hence, maternal s-DPPIV can potentially have life-long effects on the function of offspring cells and organs, which is consistent with our observations of progressive metabolic abnormalities in Off-HFD.
It is important to understand how in utero exposure to an obesogenic environment can induce such long-lasting dysregulation in offspring plasma DPPIV activity. Is there a unique factor or condition that mediates this process? In the setting of DIO, increased DPPIV production and shedding by metabolic cells such as hepatocytes and adipocytes is driven by ER stress and NFkB pathways Reference Ghorpade, Ozcan and Zheng63 . Maternal obesity is reportedly associated with signs of increased ER stress and activation NFkB-driven pathways in offspring cells Reference McCurdy, Schenk and Hetrick71 , which provides a partial explanation for our findings. It remains to be discovered how exposure to maternal obesity affects DPPIV production in different offspring cell populations, and how such a process could be diametrically opposite between male and female offspring.
Converging evidence strongly suggests that when exposed to adverse gestational conditions, male fetuses are more compromised than female fetuses Reference Dunn, Morgan and Bale72,Reference Rodriguez-Gonzalez, Reyes-Castro and Bautista73 . Sex-specific differences exist at all levels of physiology, from the genomic to organ systems Reference Penaloza, Estevez and Orlanski74 , all of which could affect DPPIV production in the setting of maternal obesity. At the organ and organ systems level, possible influencing factors include variations in the neuroendocrine milieu, placental function, fetal maturation, and maternal immune reactivity. At the tissue level, possible DPPIV-inducing stimuli induce dimorphic responses in males and females, with effects such as cellular dysplasia and excessive accumulation of ROS and pro-inflammatory cytokines Reference Gambineri, Conforti and Di Nisio5 . Sex differences are also apparent at the cellular and intracellular levels Reference Penaloza, Estevez and Orlanski74 ; for example, monocytes isolated from adult offspring of obese mothers respond differently to endotoxin challenge and express different epigenetic landscapes. As discussed earlier, we posit that exposure to maternal obesity may permanently alter epigenetic landscapes in offspring stem cells, which in turn permanently dysregulate offspring DPPIV. Maternal obesity is associated with fetal sex-specific changes in the activation of epigenome-regulatory factors such as histone deacetylases (HDACs) Reference Dearden, Bouret and Ozanne75 , which can potentially increase accessibility of the DPPIV locus to transcriptional regulators such as NFkB (p50/p65) the male offspring, which is also activated in obesity, thereby increasing transcription of DPPIV mRNA and eventually production of the bioactive s- and/or m-DPPIV protein. Understanding how exposure to maternal obesity induces persistent fetal sex-dependent dysregulation of offspring plasma DPPIV activity will be critical to addressing obesity progression in the offspring of obese mothers.
The mouse model of maternal obesity studied here bears a striking phenotypic resemblance to diverse pathologies observed both in humans Reference Wankhade, Thakali and Shankar76,Reference Santangeli, Sattar and Huda77 and in animal models of maternal obesity Reference Taylor, Matthews, Khan, Rees, Itani and Poston78 . One of our novel findings is that Off-RD and Off-HFD mice are similar in terms of food consumption and energy expenditure, which suggests that the Off-HFD do not develop obesity because of ‘obesogenic’ lifestyle choices such as overeating and/or having insufficient physical activity. In addition, our observation that exposure to maternal obesity leads to metabolic dysfunction (increased adiposity, glucose intolerance, and insulin resistance) more so in male offspring than in their female siblings, along with the sexual dimorphism in maternal and offspring activation of DPPIV, provides further evidence of sex differences in developmental programming72,73. Several prior studies have demonstrated sexual dimorphism in human offspring of obese mothers. Mingrone et al. Reference Mingrone, Manco and Mora79 reported sex-dependent metabolic changes, with males showing significantly larger fat mass, higher levels of blood insulin, and higher total insulin secretion. A longitudinal analysis of children of obese mothers also reported increased body fat at 2-6 years of age in males but not in females Reference Andres, Hull, Shankar, Casey, Cleves and Badger80 . The Helsinki Birth Cohort Study Reference Eriksson, Sandboge, Salonen, Kajantie and Osmond81 , which followed participants born in 1934–1944 and conducted follow-ups from 1971 to 2010, found sex-related differences in terms of the occurrence of coronary heart disease, type 2 diabetes, and stroke. Notably, the association of maternal BMI with offspring coronary heart disease was statistically significant only in males, whereas the association of maternal BMI with stroke was significant only in females. It appears possible that female offspring are more protected from the metabolic consequences of maternal obesity. For one, estrogen and its receptors have been found to be protective against obesity, type 2 diabetes, and CVD Reference Morselli, Santos, Criollo, Nelson, Palmer and Clegg82 . Estrogens moreover have significant effects on insulin sensitivity and on the body’s response to changes in glucose levels, two key factors mediating the programmed effects of maternal obesity Reference Clegg, Riedy, Smith, Benoit and Woods83,Reference Sandoval, Ertl, Richardson, Tate and Davis84 . Actions of estrogens therefore could at least partially explain the sexual dimorphism observed in susceptibility to early-life programming and increase in DPPIV activity in 11-month-old female offspring.
Expanding on the role of glucose, DPPIV inhibitors such as sitagliptin are effective against T2DM Reference Lee, Jang, Song, Kim and Kim11–Reference Sarkar, Nargis, Tantia, Ghosh and Chakrabarti13,Reference Williams, Vieira De Ribeiro and Prakoso15 . Sitagliptin improved glycemic control in randomized controlled clinical trials involving adults with T2DM, indicated by a 20% reduction in hemoglobin A1C Reference Yoshikawa, Tsuchiya and Kido22–Reference Tang, Huang, Liu, Shankar, Ganz and Rajpathak27 ; this protective effect has been attributed to decreased degradation of the incretin hormones GLP-1 and GIP, both of which potentiate glycemic control after a meal and promote satiety, acting over time to improve metabolic function Reference Mulvihill and Drucker62 . However, the effects of sitagliptin on weight control remain unclear. Initial clinical reports indicate that sitagliptin is “weight neutral” in adults with T2DM Reference Mulvihill and Drucker62 ; however, in most such studies, the subjects have an advanced form of obesity involving co-morbidities such as insulin resistance that are known to respond poorly to weight-loss strategies. In contrast, some clinical reports along with studies in DIO mice support that sitagliptin therapy can promote weight loss Reference Ferjan, Janez and Jensterle29,Reference Seck, Engel and Williams-Herman33 . Ultimately, it remains unclear whether sitagliptin can effectively reverse the progression of obesity and associated metabolic disorders.
In mice, increased DPPIV production in the absence of an obesogenic stimulus such as a HFD has been shown to lead to spontaneous development of obesity Reference Conarello, Li and Ronan60 , indicating that increased DPPIV signaling can independently cause obesity. Inhibiting the catalytic activity of DPPIV can reverse this effect, which is consistent with our findings that chronic sitagliptin therapy can delay obesity progression in Off-HFD males. Furthermore, our data revealed that maternal inhibition of DPPIV improves offspring metabolic profiles, proving our hypothesis that maternal DPPIV plays a key role in developmental programming. It has been previously reported that maternal sitagliptin administration improves glucose metabolism in male offspring of HFD-fed mother Reference Zhang, Xiao and Zheng85 , and our data supports this finding. However, in contrast to this report, we found no effect of maternal sitagliptin on insulin sensitivity in offspring. Different findings might be explained by selection of the animal model (rats vs. mice), duration of maternal HFD feeding prior to conception (4 weeks vs. 8 weeks), routes of drug administration (oral gavage vs. drinking water), or sitagliptin dosage (10 mg/kg vs. 0.3 mg/kg). Lack of effect of maternal sitagliptin on insulin sensitivity in male offspring suggests potentially complex role for DPPIV in the establishment of glycemic control mechanisms during gestation – a process that likely involves the development of the fetal liver, pancreas, skeletal muscles, and adipose tissue. Understanding how DPPIV regulates developmental programming is an area of future investigation that will be both important and fascinating to pursue and will be critical in our efforts to combat the pandemic of obesity and chronic disease.
In summary, it is clear that the environment during fetal and early neonatal life is a major determinant of health throughout the lifespan. Therefore, adverse early-life conditions such as exposure to maternal obesity must be considered in biomedical research and in clinical settings. As of 2018, more than half of pregnant mothers in the US are either overweight or obese Reference Yoshida, Kimura, Noda, Takeuchi and Kawakami86 ; it is thus likely that their offspring will be programmed to develop obesity. To halt the progression of this looming healthcare crisis, it is important to understand the etiologies of the various forms of obesity, including that induced by exposure to maternal obesity. Our findings highlight a role for DPPIV in the transgenerational propagation of obesity from obese mothers to their children, suggest that dysregulation of plasma DPPIV activity in pregnant mothers or in their newborn children can serve as a marker of developmental programming, and justify clinical trials using DPPIV inhibitors to address the adverse consequences of maternal obesity.
Materials and methods
Study approval
Human maternal and cord blood samples were collected from labor and delivery units at the Oregon Health & Science University under protocol approved by the Institutional Review Boards and with informed consent from the patients. All animal experiments were approved by the Oregon Health & Sciences University’s Institutional Animal Use Committee.
Methods
Extended descriptions of methods, sources of materials, and data analysis, are provided in Supporting Information.
Statistics
Means were compared using 2-way ANOVA and student’s t-test (corrected for multiple comparisons). Error bars reflect standard errors of the mean (SEM). Means were compared using two-way analysis of variance (2-way ANOVA) and student’s T-test (corrected for multiple comparisons), and results of both tests are indicated in each respective graph. Significance was set at an alpha of 0.05, which when relevant, was indicated by a symbol in each respective graph. PInt=2-way ANOVA Interaction p value.
Supplementary material
For supplementary material accompanying this paper visit https://doi.org/10.1017/S2040174422000010
Acknowledgments
We thank Yem Alharithi for technical assistance, and laboratory of Daniel Marks, MD, PhD, for giving us an access to EchoMRI. We thank OHSU Division of Comparative Medicine, Physiology and Pharmacology Graduate Program, and Vanderbilt University Mouse Metabolic Phenotyping Core (MMPC).
Author Contributions
KRCM and AM conceived and planned experiments. KRCM, MB, and EP conducted the experiments, MB and EP collected human samples, CL, JB, ES, PK, and PWN provided samples from non-human primates. SR contributed to the interpretation of the micro-CT results. KRCM and AM wrote the manuscript. All authors provided feedback and assisted with preparing final manuscript.
Financial support
This work was provided by the NIH HD099367 (AM) and NIDDK Mouse Metabolic Phenotyping Centers (National MMPC, RRID:SCR_008997, www.mmpc.org) under the MICROMouse Program, grants DK076169 (AM).
Conflicts of interest
The authors declared no conflict of interest.
Ethical standards
The authors assert that all procedures contributing to this work comply with the ethical standards described in the Public Health Service (PHS) Policy on Humane Care and Use of Laboratory Animals and the Guide for the Care and Use of Laboratory Animals and have been approved by the Oregon Health & Sciences University’s Institutional Animal Use Committee (IACUC Protocol IP0432). Human samples were collected from labor and delivery units at the Oregon Health & Science University under protocol approved by the Institutional Review Board (IRB ID STUDY00017798), and with informed consent from the patients.













 Open access
Open access