


INTRODUCTION
Papillomaviruses are members of the Papillomaviridae family [Reference de Villiers1]. To date, over 100 human papillomavirus (HPV) types have been identified [Reference Bernard2]. The viral genome is an ~8000-bp molecule of double-stranded circular DNA [Reference de Villiers1]. The genomic organization comprises three main regions: early, late and the long control region (LCR) [Reference Howley, Lowy, Knipe and Howley3]. The early region encodes for the regulatory proteins (designated E1–E7) associated with replication, transcription and cell cycle control while the late region encodes for the structural proteins. The LCR, also referred to as the upstream regulatory region (URR), is a non-coding region that includes a variety of transcriptional regulatory motifs, the early promoter and viral coded E2 regulator binding sites. It extends from the termination of the L1 gene to the first methionine of the E6 gene [Reference Howley, Lowy, Knipe and Howley3].
Recurrent respiratory papillomatosis (RRP) is caused by HPV infection of the respiratory tract, usually the larynx. It is commonly associated with HPV types 6 and 11, with most studies finding HPV-11 disease to be more aggressive than HPV-6 disease [Reference Rabah4–Reference Seedat6]. Molecular characterization has indicated that HPV-6 isolates can be divided into three variants: HPV-6a, HPV-6b and HPV-6vc while HPV-11 isolates can be divided into two genetically distinct variant groups [Reference Heinzel7, Reference Maver8].
The complete genomes of HPV-6b and closely related HPV-6a and HPV-6vc have been cloned and fully characterized. Isolates are grouped into prototypic HPV-6b-related and non-prototypic HPV-6a/6vc-related genomic variants based on their nucleotide sequences [Reference Heinzel7]. Each region of the HPV-6 genome appears to have characteristic mutations that can be used to characterize HPV-6 isolates as prototypic or non-prototypic, although the E6 and E5a regions do not appear to be able to clearly distinguish HPV-6a and HPV-6vc [Reference Kocjan9]. The non-coding region of HPV-6 and HPV-11 is a variable region with small deletions and insertions, probably the result of errors in viral DNA synthesis. This variability makes it a useful region for identifying genetic diversity. Although to date an association between genomic variants and clinical disease has not been shown, there is evidence suggesting that mutations, changes and rearrangements in the LCR are associated with E6/E7 expression in HPV-16- and HPV-18-associated genital malignancies [Reference Lace10]. Similarly, duplications and rearrangements in the URR have been found in HPV-6- and HPV-11-associated malignancies, although functional assays of the LCR have to date shown no major differences in the activities of the early promoters [Reference Kocjan9, Reference Chin, Broker and Chow11, Reference Rübben12]. Molecular characterization of HPV genomes circulating in patients with RRP and functional assays should help to determine the significance of mutations in the course of disease.
There is currently no information available regarding the genetic diversity of HPV-6 variants circulating in South Africa. Hence we considered it necessary to determine the variants in our cohort of patients with RRP. The aim of this study was to determine the HPV-6 variants affecting our patients and to determine whether mutations, duplications, insertions and rearrangements in the URR are associated with disease aggression and progression.
MATERIALS AND METHODS
Of the 31 patients who underwent surgery for RRP at Universitas Academic Hospital, a government training hospital in Bloemfontein, South Africa, between May 2008 and May 2010, 18 had disease due to HPV-6 while in 13 the disease was due to HPV-11. Seventeen of the patients were newly diagnosed with RRP in the treatment period while 14 were known patients with RRP. Twelve of the patients with HPV-6 disease were included in this study.
Severity of disease was determined at each surgical intervention using the Derkay staging system [Reference Derkay13] and an average of Derkay scores over the total treatment period at our institution was used for the data analysis. This staging system has been demonstrated to have a high level of inter-observer reliability in staging of the disease [Reference Hester14]. The average number of procedures per year was only calculated for patients who had undergone at least four procedures or who had at least 6 months of follow-up data. Patient VBD 77/09 had onset of disease in childhood but was previously treated at another hospital. Her clinical records were only available from 1990, when treatment began at our institution. Clinical data was analysed and summarized at the end of December 2010 since all patients had on-going active disease.
To identify HPV-6 variants based on genome mutations or changes, the non-coding region of the genome and the gene encoding the E6 protein were amplified using primers as previously described: forward primer designated HPV6 – LCRForward (5′-CTG CTG TTT CCA AAG CCT CT-3′), genomic position relative to HPV-6b 7233-7252 and reverse primer designated HPV6 – E6Reverse (5′-CCA CTT CGT CCA CCT CAT CT-3′), genomic position relative to HPV-6b 650-631 [Reference Seedat6]. The primer pair amplify a region of the genome that includes the predicted full length of the LCR genomic region, 712-822 bp, and the complete E6 open reading frame (ORF) (453 bp) of the HPV-6 genome. In order to obtain a complete sequence, primer walking was performed with internal primers which were identified with the use of sequence data retrieved from GenBank for HPV-6a (L41216), HPV-6b (NC001355, X00203) and HPV-6vc (AF092932). These sequences were aligned with partial sequence data obtained from this study which resulted in the following forward primer designated HPV6-F LCRint (5′-CAC CCT GTG ACT CAS TGG CTG-3′), genomic position relative to HPV6b 7429-7449 and the following reverse primer designated HPV6-R E6int (5′-CGT ATG CAT AGA TAG ATT AAA CGT CTT G-3′), genomic position relative to HPV6b 179-152. Alignments were performed with the use of Clustal X version 2.0.11.
Laryngeal biopsies were submitted to the laboratory and processed immediately or frozen at −20°C until processed. Total DNA was extracted from biopsy material using QIAamp DNA mini kit (Qiagen Inc., USA) according to the manufacturer's instructions. PCR amplifications of HPV-6-positive samples were performed using the Expand High Fidelity DNA PCR system kit (Roche Applied Science, Germany) according to manufacturer's instructions. Briefly, the reaction mix comprised 0·3 μm of each primer, 0·2 mm dNTP mix, 2·6 units Expand High Fidelity enzyme, reaction buffer and 1–10 ng DNA template. The reactions were cycled on a Gene Amp® PCR system instrument model 9700 using the following cycle conditions: denaturation at 94°C for 2 min and 30 cycles of 94°C for 15 s, 47°C for 30 s, 72°C for 1 min, a final elongation step was performed at 72°C for 7 min and the tubes were held at 4°C until removed. PCR products were separated by electrophoresis through a 1% agarose gel stained with ethidium bromide and visualized under UV light. The predicted size of the amplicon was about 1320-1430 bp. PCR products were purified from the agarose gel for downstream applications using the Promega Wizard® SV Gel PCR Clean-UP System kit (Promega, USA) according to manufacturer's instructions.
The nucleotide sequences of the amplicons were determined using Big Dye™ Terminator Sequencing Reaction kit with Amplitaq DNA polymerase FS (Applied Biosystems, USA) according to the manufacturer's instructions and each of the following primers described above: HPV6 – LCRForward, HPV6 – E6Reverse, HPV6-F LCRint and HPV6-R E6int. Sequence data were compared with data available from GenBank by performing a BLAST search analysis [National Centre for Bioinoformatics (http://blast.ncbi.nlm.nih.gov/Blast.cgi)] and subsequently aligning the data with representative data retrieved from GenBank for isolates from Slovenia. HPV-6 genomic variants were identified by alignment and analysis of nucleotide sequence data from the LCR and complete E6 genomic region and comparison with data retrieved from GenBank for HPV-6b prototype. Data were edited using ChromasPro version 1.41 and aligned using Clustal X Version 2.0.11. Sequence data retrieved from GenBank were included in order to represent each non-prototypic HPV variant: 6a and 6vc.
The study was approved by the Ethics Committee of the Faculty of Health Sciences, University of the Free State and the Clinical Head of Universitas Hospital. Written informed consent was obtained from all adult patients and the parents of children prior to their inclusion in the study.
RESULTS
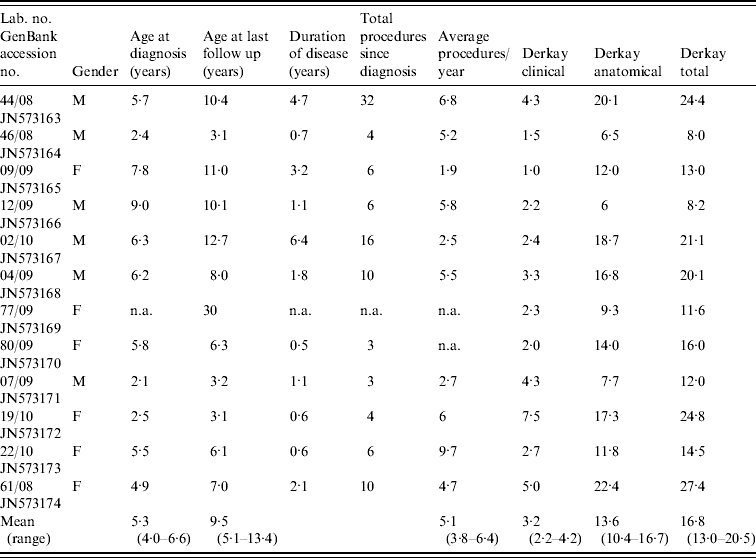
The patient cohort included 11 paediatric patients and one adult patient, who had juvenile-onset disease. The patients' details including clinical presentation and the number of surgical procedures performed on each patient during the period in which they have been treated at our facility are summarized in Table 1. None of the patients required a tracheostomy at any stage nor did any of the patients in this cohort have pulmonary metastases.
Table 1. Summary of clinical presentation for patients with recurrent laryngeal papillomatosis enrolled in the study
The complete LCR and E6 region were amplified from HPV DNA using the primers described above. The nucleotide sequence data for the LCR region and the E6 gene were edited and aligned. The genetic diversity for the non-coding region was determined using sequence data comprising 712-991 bases with variations in length a consequence of various insertions found in different isolates and a large 170-bp duplication in isolate VBD 19/10. The genetic analysis of the E6 gene was performed on 453 nucleotide bases. Sequence data retrieved from GenBank were selected to include HPV-6 isolates representative of each known variant to serve as reference for each variant and to assist with genetic identification and confirmation of variants. All subsequent identification of nucleotide positions for base changes are described relative to the prototype HPV-6b (GenBank accession no. NC001355). Sequence variations for the LCR in comparison with the prototype HPV-6b and the non-prototypic HPV-6a and HPV-6vc variants are illustrated in Figure 1 (a, b).
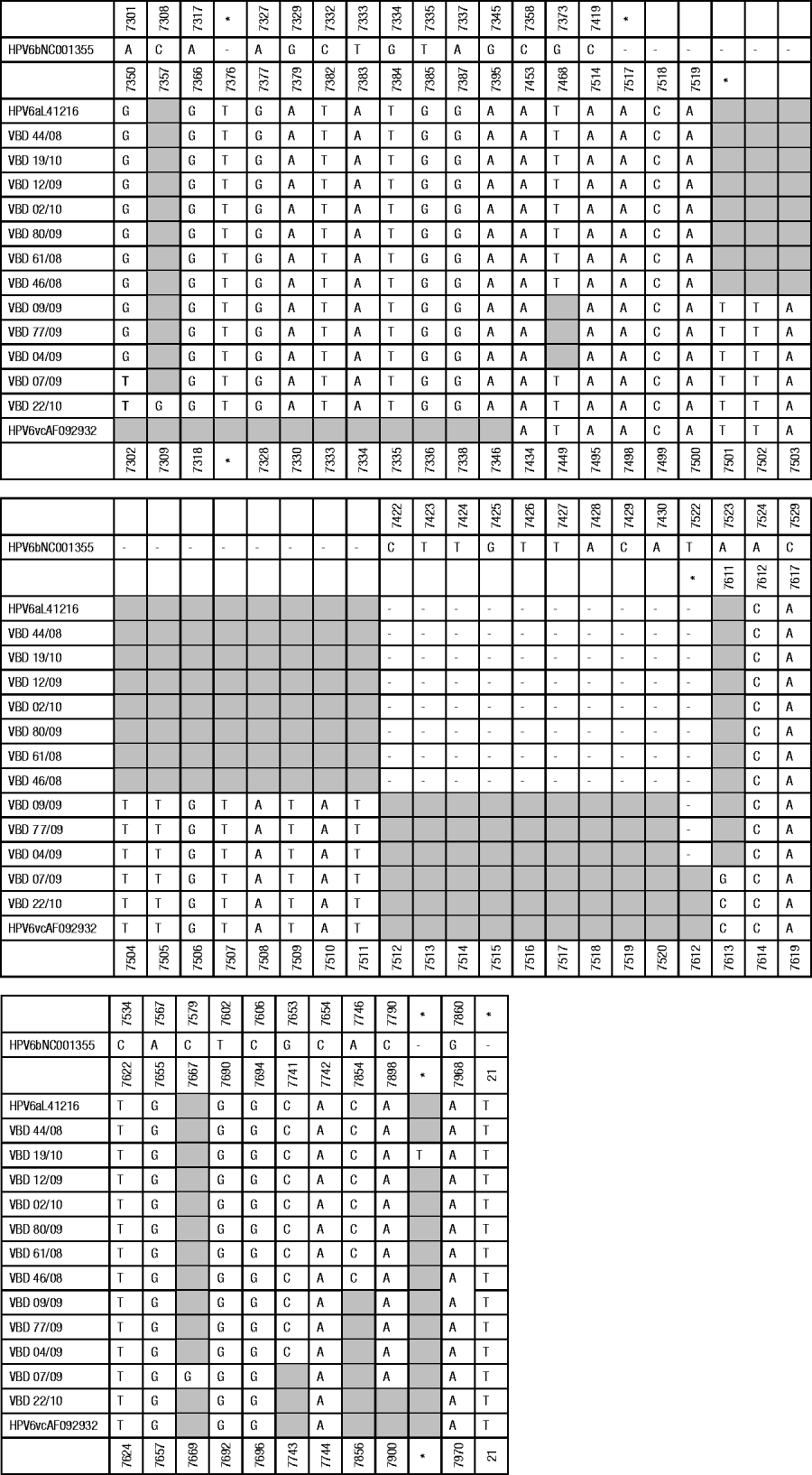
Fig. 1a. Summary of point mutations and insertions identified in the non-coding region. Base positions are relative to the reference HPV-6b (GenBank accession no. NC 001355). The 170-bp duplication in VBD 19/10 (position 7835-7836 relative to HPV-6b) has been excluded to simplify the figure. Shaded blocks represent no variation from HPV-6b. * Denotes genomic position in absence of insertion; – represents absence of bases.
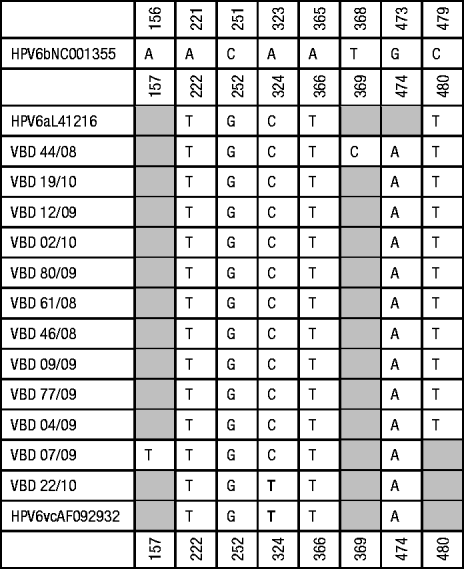
Fig. 1b. Summary of point substitutions identified in the E6 coding region. Shaded blocks represent no variation from HPV-6b. * Denotes genomic position in absence of insertion; – represents absence of bases.
The following insertions were identified by comparison with the prototype HPV-6b reference sequence: all the isolates had a 3-bp insertion between bases 7421-7422 and isolates 4/09, 7/09, 9/09, 77/09 and 22/10 had an additional 11-bp insertion at the same position, all the isolates had a T insertion between bases 7326-7327 and a T insertion between bases 20-21 and isolate 19/10 had a T insertion between bases 7835-7836. This insertion was followed by a large duplication comprising 170 bp. The region from 7774 to 7943 bp (relative to HPV-6a) was duplicated in this genome. All of the samples had a 20-bp insertion between bases 7721-7722 (not shown in Fig. 1). This insertion has previously been described as being characteristic of HPV-6a and HPV-6vc variants but is not present in HPV-6b variants [Reference Wiatrak5]. The following point mutations were identified from the sequence data: all isolates had an A–G at position 7301 except 22/10 and 7/09 which showed an A–T substitution at this position, VBD 22/10 had a C–G substitution at position 7308; isolates VBD 7/09, 22/10, 44/08, 19/10, 12/09, 02/10, 80/09, 61/08, 46/08 had a G–T substitution at position 7373; VBD 7/09 had an A–G substitution at position 7523; VBD 22/10 had an A–C substitution at position 7523; VBD 7/09 had a C–G substitution at position 7579; VBD 09/09, 77/09, 04/09, 44/08, 19/10, 12/09, 02/10, 80/09, 61/08 and 46/08 had a G–C substitution at position 7653; VBD 44/08, 19/10, 12/09, 02/10, 80/09, 61/08 and 46/08 had an A–C substitution at position 7746; VBD 09/09, 77/09, 04/09, 07/09, 44/08, 19/10, 12/09, 02/10, 80/09, 61/08 and 46/08 had a C–A substitution at position 7790. Isolates VBD 09/09, 77/09, 04/09, 44/08, 19/10, 12/09, 02/10, 80/09, 61/08 and 46/08 had a deletion at position 7522.
Based on BLAST analysis and alignment of LCR and E6 data retrieved from GenBank for 34 representative HPV-6 variants two novel variants were identified (data not shown). The following isolates, VBD 9/09, 4/09 and 77/09, had a guanine base at position 7373 (similar to HPV-6b) whereas all other HPV-6a and HPV-6c variants included in this study and retrieved from GenBank had a thymine at that position. VBD 19/10 with both an insertion and duplication in the URR genome appears to be a novel variant. Based on clinical presentation there was no evidence that VBD 9/09 and 77/09 were isolated from patients with aggressive disease but VBD 4/09 and 19/10 were isolated from patients with clinical features of severe disease (requiring >3 procedures per year and Derkay scores >20). Whether these genomic variants influence replication and expression and thereby play a role in the clinical severity of the disease will require functional studies.
The E6 gene is a highly conserved region with only eight mutations: seven synonymous changes relative to the prototype HPV-6b (most isolates had A323C substitution except VBD 22/10 which had A323T, T368C in VBD 44/08, A221T, C251G, A365T and G473A in all of the isolates, C479T in all isolates except VBD 7/09 and VBD 22/10) and a non-synonymous transition A156T in the genome of VBD 7/09 that resulted in an amino-acid change from threonine to serine. Based on BLAST searches, alignment of data from 12 South African isolates, 34 Slovenian isolates and four prototypic and non-prototypic isolates, T368C was the only novel substitution in our cohort of isolates.
DISCUSSION
Sequence data from laryngeal biopsy tissue samples were analysed and compared to genomes from prototypic HPV-6b and non-prototypic HPV-6a and HPV-6vc. A papillomavirus is recognized as a new type if the complete genome has been cloned and the DNA sequence of the L1 ORF differs by more than 10% from the closest known papillomavirus type [Reference de Villiers1]. Differences of between 2% and 10% define a subtype while differences of <2% define a variant [Reference de Villiers1]. Less conserved regions of the genome, such as the LCR region, are useful for identifying genetic variants. In the study by Kocjan et al. they reported from their results that unlike the L1, E2, E5b and LCR genomic regions, the ORFs of E6 and E5a did not provide enough information to differentiate between the HPV-6a and HPV-6vc genetic lineages [Reference Kocjan9]. The non-coding regions of HPV-6 and HPV-11 frequently differ by small deletions and insertions that may be derived from sequences that are prone to producing slippage, a misalignment-mediated DNA synthesis error, but those isolates do not differ significantly elsewhere in the viral genome [Reference Chan15]. Investigation of HPV-6 and HPV-11 sequences and their variants have implied that slippage may be a common source for producing variation in these two HPV types and that slippage over many years is responsible for the small changes in the majority of HPV-6 and HPV-11 genomes. Repetitions of G/AT in the 5′ part of the LCR make it a perfect substrate for subjection to slippage-mediated indel events [Reference Heinzel7]. High variability makes this a useful region for identifying genetic variation.
There is currently no consensus on the influence genetic mutations may have on viral replication, pathogenicity and malignant transformations. A duplication of genomic sequence in the LCR has been reported in a squamous lung carcinoma [Reference DiLorenzo16] and functional assays have suggested that duplications can have enhancer functions [Reference Wu and Mounts17]. In contrast, other functional assays have shown little or no evidence to support that mutations, insertions and duplications influence protein expression [Reference Rübben12]. It is likely that only specific changes may influence expression and pathogenicity or alternatively that various factors play a role, including host immune responses. The aim of our study was to identify variants of HPV-6 in our cohort of patients, to uncover the existence of novel mutations and variants that may exist along with the frequency of mutations and rearrangements and to compare these changes with clinical presentation in order to investigate the possibility of any correlation between changes observed and clinical presentation. Sequence data obtained from laryngeal biopsy cohort tissue samples were analysed and compared to genomes from prototypic HPV-6b and non-prototypic HPV-6a and HPV-6vc, available from GenBank. Two novel variants were identified based on the non-coding region, of which one isolate (VBD 19/10) contained both an insertion and duplication in the URR genome, with features suggestive of an aggressive clinical course. Interestingly, several of the isolates had identical sequence data to that obtained from biopsies collected from patients with benign condyloma acuminata in Slovenia suggesting that, as Kocjan et al. implied, HPV-6 variants are not geographically restricted [Reference Kocjan9].
The major function of the E6 protein in high-risk HPVs is to mediate the degradation of p53, leading to the inhibition of apoptosis in the infected host cell. The E6 genomic region has been defined as an intrinsically disordered protein, implying that small modifications can have extensive effects on its tertiary structure and function. Hence, in contrast with the non-coding region, small changes could be highly significant. The single amino-acid change in our patient infected with the novel HPV-6a may therefore prove significant in his clinical course.
In summary, collection of molecular data combined with the long-term study of our patients and in vitro functional assays should help to elucidate the molecular determinants of virulence that will have prognostic relevance, provide clarity on the role of changes in the non-coding region that may be responsible for altered expression of the E6 and E7 genes and transformation of host cells and, possibly more relevant to low-risk HPV types, determine if changes in the URR influence viral replication by influencing E2 and E1 replicating proteins as previously shown for HPV-16 [Reference Hubert18]. Functional studies will be required to confirm or exclude the influence of genetic differences on replication and expression.
ACKNOWLEDGEMENTS
This study was funded by the NHLS Research Trust and the University of the Free State.
DECLARATION OF INTEREST
None.



