


1. Introduction
DNA topoisomerases are required for altering DNA topology to facilitate replication, transcription and chromosome segregation. Eukaryotic type II DNA topoisomerases carry out these alterations in DNA structure by transiently introducing breaks in DNA via an ATP-dependent reaction and thus changes the linkage number in DNA molecules (Gellert, Reference Gellert1981; Wang, Reference Wang1985). This process reversibly decatenates double-stranded DNA (dsDNA) and modulates supercoiling that is essential for mitosis (Yanagida & Wang, Reference Yanagida and Wang1987). Top2 has been studied in detail in yeast and mammalian systems and has been suggested to be involved in cell proliferation and cell cycle regulation (Smith et al., Reference Smith, Gimenoz-Abian and Clarke2002; Wang, Reference Wang2002). In the absence of Top2, cells proceed to catastrophic mitosis, indicating the involvement of biochemical surveillance systems or checkpoints in response to the decatenatory activity of Top2 during mitosis (Downes et al., Reference Downes, Clarke, Mullinger, Gimenez-Abian, Creighton and Johnson1994). However, these checkpoints are not robust enough to maintain cell cycle arrest rather they transiently delay the cell cycle to allow some extra time for the recovery (Gimenez et al., Reference Gimenez Abian, Weingartner, Binarova, Clarke, Anthony, Calderini, Heberle-Bors, Moreno Diaz de la Espina, Bogre and De la Torre2002). Conditional mutants of budding and fission yeast lacking top2 activity at the restrictive temperature cannot form colonies because of the chromosomal breakage generated due to the mis-segregation of sister-chromatids that have not been disentangled during DNA replication (Bermejo et al., Reference Bermejo, Doksani, Capra, Katou, Tanaka, Shirahige and Foiani2007). Mammalian G2 cells treated with topoisomerase inhibitors delays entry into mitosis (Kalwinsky et al., Reference Kalwinsky, Look, Ducore and Fridland1983; Wan et al., Reference Wan, Capasso and Walworth1999) and drugs that can override checkpoints such as caffeine, bypass these delays (Downes et al., Reference Downes, Clarke, Mullinger, Gimenez-Abian, Creighton and Johnson1994). Thus, it was suggested that this effect was due to a G2 checkpoint that monitors Top2 activity. These inhibitors induced G2 delays without directly causing DNA strand breaks (Tanabe et al., Reference Tanabe, Ikegami, Ishida and Andoh1991; Downes et al., Reference Downes, Clarke, Mullinger, Gimenez-Abian, Creighton and Johnson1994). Recently, it has been suggested that these inhibitors may induce DNA damage due to interrupted decatenation, indicating an overlap between G2 DNA damage and Top2 checkpoints (Dominguez et al., Reference Dominguez, Pastor, Mateos and Cortes2001; Mikhailov et al., Reference Mikhailov, Cole and Rieder2002; Hajji et al., Reference Hajji, Pastor, Mateos, Dominguez and Cortes2003; Adachi et al., Reference Adachi, Iiizumi, So and Koyama2004). Studies with temperature sensitive top2 mutants in budding and fission yeast suggest that Top2 plays an important role in decatenating sister chromatids before anaphase (DiNardo et al., Reference DiNardo, Voelkel and Sternglanz1984; Uemura & Yanagida, Reference Uemura and Yanagida1984; Holm et al., Reference Holm, Goto, Wang and Botstein1985) but their involvement in mitotic checkpoint is still elusive. CPT, a topoisomerase 1 inhibitor activates Chk1 and induces a DNA damage checkpoint response in fission yeast (Wan et al., Reference Wan, Capasso and Walworth1999). Recently, it has been proposed that compromised Top2 function activates a checkpoint system that relies on a subset of spindle checkpoint protein rather than DNA damage checkpoint. However, this checkpoint system is operated by a novel mechanism that is independent of the pds1/securin pathway (Andrews et al., Reference Andrews, Vas, Meier, Giménez-Abián, Díaz-Martínez, Green, Erickson, VanderWaal, Hsu and Clarke2006). In the present study, we have demonstrated a conditional synthetic lethality of a Top2 mutant with DNA damage checkpoint kinase Chk1. In the absence of Chk1-dependent checkpoint response top2 mutant cells fail to delay mitotic progression and exhibit chromosome segregation defects. Phosphorylation of the checkpoint protein kinase Chk1 takes place in the presence of defective top2 activity during mitosis and this activation of Chk1 requires cells to pass through the G2 phase of the cell cycle. Furthermore, treating cells with microtubule poison (TBZ) partially increases the survival of top2–10 mutant cells at a non-permissive temperature.
2. Materials and methods
(i) Strains and growth condition
Schizosaccharomyces pombe strains used in this study are listed in Table 1. Standard genetic methods were utilized for making strains as described in Moreno et al. (Reference Moreno, Klar and Nurse1991) . For temperature-shift experiments, cells were grown to mid-log phase at 25°C and then shifted to restrictive temperature 34°C in a water bath. To assess checkpoint arrest, cells were synchronized in S phase by growing them in 12 mM hydroxyurea (HU) for 4 h at 25°C, washed and released at 34°C. The samples were collected at a 30 min interval and the fraction of binucleate or divided cells was determined as an indication of entry into mitosis using 4′,6′ diamidino-2-phenylindole (DAPI) staining as described previously (Ahmed et al., Reference Ahmed, Palermo, Wan and Walworth2004).
Table 1. Strains used in this study

(ii) Identification of gene by complementation analysis
In order to identify the gene coding for ts10/top2–10 mutant, the S. pombe genomic DNA library was transformed in ts10/top2–10 mutant strain by lithium acetate method (Moreno et al., Reference Moreno, Klar and Nurse1991). Leu+ Ts+ transformants were obtained and plasmid was recovered from Leu+ Ts+ co-segregants. Restriction digestion and sequencing analysis identify ~15·4 kb region containing two open reading frames. The 4 and 8 kb HindIII fragment coding for apc10 and top2 genes, respectively, were further sub-cloned in psp1 vector and re-transformed in ts10/top2–10 mutant strain for complementation analysis.
(iii) Microscopy
For staining nucleus, cells were grown to mid-log phase at 25°C then shifted to 34°C, samples were collected at indicated times, fixed with 70% ethanol and stained with DAPI and/or calcofluor, visualized using a fluorescence microscope. About 200–300 cells were counted for aberrant nuclei and percentage was plotted.
(iv) Preparation of lysates and Western-blot analysis
Strains with a triple-haemagglutinin tagged allele of chk1 integrated at the genomic chk1 locus (chk1:ep) were used (Walworth & Bernards, Reference Walworth and Bernards1996). Cells were grown to mid-log phase at 25°C then shifted at 34°C in water bath for different time periods. Cells were harvested by centrifugation and lysed using glass beads and a vortex machine. Lysate in phosphate-buffered saline was centrifuged at 5000 rpm in a microfuge for 5 min at 4°C. Supernatant was collected and protein estimation was performed using the Bradford assay method. For Western-blot analysis, about 200 μg of total cell lysate was run on 8% SDS-PAGE, transferred to nitrocellulose membrane and probed with F7 (Santa Cruz) or 12CA5 antibody as described earlier (Walworth & Bernards, Reference Walworth and Bernards1996). A peroxidase-coupled secondary antibody and the enhanced chemiluminescence detection system (GE lifesciences) were used to detect the immune complexes.
(vi) Cloning of mutant top2 gene by the gap repair method
Mutant topoisomerase gene was cloned by the gap repair method. Wild-type top2 gene cloned in psp1 vector was digested with StuI–SphI restriction enzyme that deleted 1412 amino acids of top2 coding region. Then 10·7 kb linear fragment containing upstream and downstream sequences of top2 gene with a leucine selectable marker was isolated by gel purification and transformed in top2–10 mutant strain. Transformants containing plasmid with mutant allele of top2 gene was distinguished from those with integrated plasmid by the mitotic stability of leucine marker present on the plasmid. Plasmid containing mutant copy of top2 gene was recovered from the transformant as described in Moreno et al. (Reference Moreno, Klar and Nurse1991), and was confirmed with HindIII digestion. The complete gene coding for top2 was sequenced using appropriate primers and was compared with the wild-type sequence of top2 gene.
3. Results and discussion
(i) Identification of mutation leading to synthetic lethality in combination with chk1 deletion
In order to identify conditional lethal mutants, the chk1 knockout strain was mutagenized by ethyl methanesulphonate (EMS). Mutant cells that do not form colonies at a non-permissive temperature (36°C) were subsequently isolated. Thirty-seven such mutants were checked for their inability to grow at the semi-permissive temperature (33°C). Five such mutants that were unable to form colonies at 33°C were backcrossed and their ability to grow at 33°C in wild-type and chk1 knockout background was compared. One such mutant (ts10/top2–10) was able to grow at 33°C but in chk1 deletion background the cells were unable to form colonies (Fig. 1(a)), indicating a conditional lethality of ts10/top2–10 mutant with chk1 knockout. Even at the permissive temperature (25°C) the ts10/top2–10 chk1 deletion strain exhibits slower growth compared to the ts10/top2–10 single-mutant strain (Fig. 1(a)), indicating a more severe defect in the double mutant as compared to the single mutant. To further confirm the conditional lethality associated with the chk1 knockout, we checked the ability of ts10/top2–10 and chk1 delete ts10/top2–10 double-mutant strain to form colonies at the permissive temperature after transiently exposing them to a non-permissive temperature (34°C). The colony-forming ability of chk1 delete ts10/top2–10 double mutant is less as compared with the ts10/top2–10 single mutant after exposing the cells at 34°C for a short period of time (Fig. 1(b)). The inability of the ts10/top2–10 mutant to form colonies in the chk1 knockout background indicates that in the absence of normal checkpoint response the ts10/top2–10 mutant cells lose viability at a faster rate compared with the cells exhibiting proficient checkpoint response.

Fig. 1. The ts10/top2–10 mutant allele shows conditional lethality with chk1 knockout: (a) Indicated strains were streaked on YEA plates and incubated at indicated temperature for 2–4 days before taking photograph. (b) Per cent survival in top2–10 chk1 knockout strain at a non-permissive temperature: indicated strains were grown till mid-log phase at 25°C then shifted at 34°C. Sample were collected at 2 h intervals, equal number of cells were plated on YEA plates and incubated at 25°C. Number of surviving colonies was calculated as the percentage of colonies appearing at permissive temperature. Values shown are the average of three independent experiments with standard deviation. (c) Per cent aberrant mitosis in top2–10 mutant cells at non-permissive temperature: indicated strains were grown till mid-log phase at 25°C and then shifted at 34°C. Cells were collected at the indicated time interval, fixed with 70% ethanol and stained with DAPI. Cells having aberrant chromosome segregation were counted.
(ii) Identification of gene coding for ts10/top2–10 by complementation analysis
The gene coding for the ts10/top2–10 mutation was identified by complementation analysis as described in section 2. Sequencing and database analysis identified a 15·4 kb region containing two open reading frames coding for top2 and apc10 genes, respectively (Fig. 2(b)). Further sub-cloning and complementation analysis identified top2 as the ts10 coding gene, while the apc10 gene could not complement the temperature-sensitive phenotype of the ts10/top2–10 mutant (data not shown).
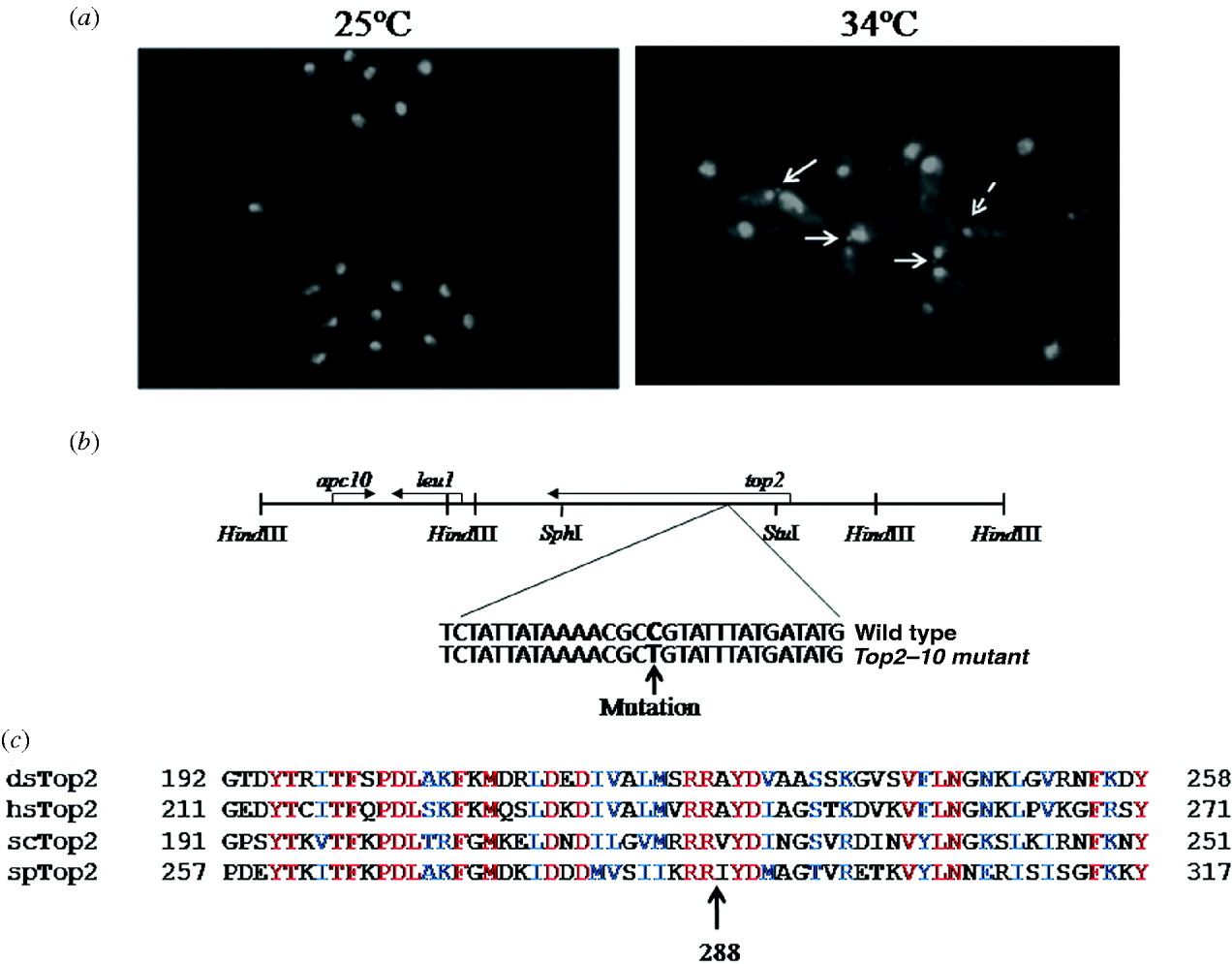
Fig. 2. (a) top2–10 mutant cells exhibit chromosome segregation defect at a non-permissive temperature: cells were grown at 25°C till mid-log phase and then shifted at 34°C for 4 h, fixed with 70% ethanol and stained with DAPI. Arrow indicates defective chromosome segregation. (b) Schematic map of 15·4 kb region of plasmid (upper panel), indicating 8·0 and 4·0 kb HindIII fragment containing top2+ and apc10+ genes, respectively. Location of mutation in top2 gene has been shown in the lower panel. (c) Multiple alignment of Top2 from S. pombe (spTop2; accession no. P08096), Drosophila (dmTop2; accession no. P15348), S. cerevisiae (scTop2; accession no. P06786) and humans (hsTop2; accession no. P11388) are shown. The conserved arginine residue at position 288 in S. pombe is marked.
(iii) The ts10/top2–10 mutant exhibits chromosome segregation defects at the non-permissive temperature
To further investigate the defective phenotype in the top2–10 mutant, we grew cells at 25°C and then transferred them to 34°C for 6 h. DAPI staining was subsequently performed using a fluorescence microscope. In top2–10 mutant cells, nuclear chromatin region was not properly segregated but the cells were separated due to the cytokinesis (Fig. 2(a)). About 21% top2–10 mutant cells exhibit these chromosome segregation defects after 6 h at the non-permissive temperature compared to wild-type cells that did not exhibit segregation defects under the same growth conditions (Fig. 2(a)). Additionally, a number of cells in top2–10 mutant were having single nuclei at the edge of the cells (Fig. 2(a), dotted arrow). These nuclei could have been generated due to the separation of two daughter cells having aberrant mitosis. Consistently different mutant alleles of top2 have also been shown to exhibit similar kinds of chromosome segregation defects (Uemura & Yanagida, 1984, Reference Uemura and Yanagida1986).
(iv) Combination of top2–10 with the chk1 deletion aggravates the segregation defects
The lethality of the top2 mutant in chk1 deletion background at a semi-non-permissive temperature prompted us to look for mitotic defects in the chk1 knockout and top2–10 double-mutant strains. Wild-type, chk1 delete, top2–10 mutant and top2–10 chk1 delete strains were grown at permissive temperature (25°C) till the mid-log phase and then shifted to a non-permissive temperature (34°C). Samples were taken at regular time intervals. Wild-type and chk1 delete cells exhibit normal chromosome segregation, while about 78% top2–10 chk1 delete cells exhibit chromosome segregation defects during mitosis as compared with top2–10 single mutant that has only about 34% chromosome segregation defects after 4 h at 34°C (Fig. 1(c)). During the same period of time there was considerable reduction in survival pattern of top2–10 and chk1 delete top2–10 cells (Fig. 1(b)). Further incubation at non-permissive temperature results in about 75% aberrant mitosis in top2–10 mutant as compared to 89% in double mutant. These results strengthen our observation that in the absence of a Chk1-dependent checkpoint response, top2–10 mutant cells exhibit increase in chromosome segregation defect.
(v) Mapping and identification of top2–10 mutation by gap repair
Furthermore, to identify the mutation in ts10/top2–10 gene we cloned the mutant top2 gene by gap repair as described in section 2. Transformation of plasmid containing mutant copy of top2 gene in top2–10 mutant strain does not suppress the temperature sensitive phenotype of the mutant strain (data not shown), suggesting that the isolated plasmid indeed contain mutant copy of top2 gene. Sequencing and comparison with wild-type sequence of top2 gene indicated a mutation from C to T, which changes Arg 288 to Cys (Fig. 2(c)). The arginine residue at 288 is conserved across the species (Fig. 2(c)) indicating that this residue might be having some important role in topoisomerase function. Furthermore, these results also rule out the possibility that top2 could be a suppressor of ts10 mutant strain.
(vi) Topoisomearse 2 mutant cells mildly delay the mitotic progression at a non-permissive temperature that required the checkpoint kinase protein Chk1
In order to investigate the role of top2–10 mutant in Chk1-dependent checkpoint function, we examine mitotic entry at non-permissive temperature in top2–10 mutant cells. The top2–10 mutant cells delay mitotic entry at a non-permissive temperature (Fig. 3(a)). More importantly, top2–10 mutant cells fail to delay mitotic entry in a chk1 deletion background (Fig. 3(a)), suggesting that the genotoxic stress response in top2–10 mutant cells at a non-permissive temperature generate a Chk1-dependent checkpoint response.

Fig. 3. Topoisomerase 2 mutant cells mildly delay the mitotic progression and activates checkpoint kinase Chk1: (a) cells were synchronized in S phase by growing them in 12 mM HU for 4 h at 25°C, washed and released at 34°C, samples were collected at 15 min interval and the fraction of binucleate or divided cells was determined as an indication of entry into mitosis using DAPI staining. (b) Indicated strains were grown at permissive temperature till mid-log phase then shifted at 34°C for indicated time. Protein lysate was prepared as described in Materials and methods, samples were run on 8% SDS-PAGE, transferred on nitrocellulose membrane and probed with anti-HA antibody. (c) Per cent survival in top2–10 mutant cells arrested in G2: indicated strains were grown till the mid-log phase at 25°C then shifted at 34°C. Samples were collected at a 2 h interval; equal number of cells were plated on YEA plates and incubated at 25°C. Number of surviving colonies was calculated as the percentage of colonies appearing at permissive temperature. Values shown are the average of three independent experiments with standard deviation. (d) Per cent survival in top2–10 mutant cells grown in the presence of TBZ: indicated strains were processed as described in (c) except that the cultures were shifted at 34°C in the presence or absence of 30 μg/ml TBZ.
(vii) Topoisomerase 2 mutant cells activate the DNA damage checkpoint protein kinase Chk1 during mitosis
In response to DNA damaging agents, the Chk1 protein gets phosphorylated due to the activation of checkpoint and hence migrates slowly on SDS-PAGE (Walworth & Bernards, Reference Walworth and Bernards1996). Reduced viability in top2–10 chk1 deletion background and delay in mitotic progression in top2–10 mutant cells prompted us to question whether the top2–10 mutant allele also activates Chk1. To ascertain the activation of Chk1, we introduced the HA-tagged Chk1 in the top2–10 mutant background by genetic crosses. Chk1 was monitored by Western blotting probed with anti-HA antibody (F7). As shown in Fig. 3(b), the slow migrating form of Chk1 starts appearing in top2–10 mutant cells after 3 h and becomes more prominent after 6 h at a non-permissive temperature (34°C) indicating phosphorylation of Chk1 in top2–10 mutant cells, while the wild-type cells grown under same conditions do not exhibit this phenomenon (Fig. 3(b), upper panel). This result clearly suggests that defects in Top2 activity leads to the activation of the checkpoint kinase protein Chk1.
(viii) Activation of the Chk1 kinase in topoisomerase mutant is a post-G2 event
The protein kinase Chk1 is phosphorylated in response to a variety of DNA-damaging agents, such as UV light, topoisomerase I poison camptothecin (CPT) and inactivation of DNA ligase (Walworth & Bernards, Reference Walworth and Bernards1996; Wan et al., Reference Wan, Capasso and Walworth1999). A DNA damage signal could be generated in cells harbouring mutation in top2 gene by one of at least two mechanisms. In one scenario, the double-strand break can be generated in response to defective topoisomerase 2 functions at the time of DNA replication. Alternatively, in top2 mutants the attempt to segregate entangled chromatids could cause chromosomal breakage at the time of mitosis; a process that might also activate the Chk1 kinase. To check these possibilities a conditional mutant cdc25–22 that arrests cell cycle in G2 was used. As shown in Fig. 3(b), top2–10 mutant cells that were arrested in G2 failed to activate Chk1 when shifted to a non-permissive temperature (34°C). This suggests that in top2 mutants the activation of Chk1 takes place only if cells try to enter into mitosis. Furthermore, the survival pattern of the cdc25–22 top2–10 double mutant was better as compared to the top2–10 single mutant at 34°C (Fig. 3(c)). Consistent with this the cdc25–22 top2–10 double-mutant cells were as long as cdc25–22 single mutants with single nuclei (data not shown), suggesting an arrest in G2 phase of the cell cycle. Furthermore, there was partial improvement of survival at 34°C in the top2–10 mutant cells grown in the presence of microtubule poison (TBZ) as compared with the cells grown in its absence (Fig. 3(d)), suggesting that decrease in viability in the top2–10 mutant cells could be due to the catastrophic mitosis. There was reduction in viability in wild-type cells treated with TBZ that could be due to some toxic effect of the drug (Fig. 3(d)). This could explain why there was only marginal improvement of survival in top2–10 mutant in the presence of TBZ as compared to a robust increase in survival in cdc25–22 top2–10 mutant cells (compare Fig. 3(d) with Fig. 3(c)).
(ix) Analysis of cell cycle kinetics in synchronized top2–10 mutant
To monitor the cell cycle progression, we synchronized the cells in S phase by growing them in the presence of 12 mM HU at a permissive temperature for 4 h; the cells were washed and released at a non-permissive temperature (34°C). During the first cell cycle, the septation index was high at 90 min time points indicating a normal cytokinesis. More importantly, in wild-type and chk1 delete cells, the septation index starts increasing after 165 min indicating normal second cell cycle progression. In contrast to this, the septation index remained low in top2–10 and top2–10 chk1 delete cells, suggesting that these cells were unable to enter into the next cell cycle (Fig. 4(a)), possibly due to the catastrophic defects just after the first mitosis. There was 30–45 min delay in mitotic entry at a non-permissive temperature in top2–10 mutant as compared to the top2–10 chk1 delete cells (Fig. 3(a)). Consistently, only 4% of top2–10 cells had aberrant mitosis after 75 min shift at a non-permissive temperature as compared to 32% in top2–10 chk1 delete cells (Fig. 4(b)), indicating the involvement of Chk1-dependent checkpoint response in top2–10 mutant cells at the time of mitosis. Additionally, the per cent survival of top2–10 mutant and top2–10 chk1 delete mutant cells decreases as the percentage of aberrant mitosis increases (Fig. 4(c)). To determine the timing of the activation of Chk1 in top2–10 mutant cells, the Chk1 phosphorylation was monitored in synchronized population after releasing from HU arrest. As shown in Fig. 4(d), a small fraction of Chk1 was phosphorylated in HU-arrested cells in top2–10 mutant cells. The level of phosphorylation in top2–10 mutant cells decreases in 1 h coincidentally the percentage of aberrant mitosis was also negligible at the same time point (Fig. 4(d)). The fraction of phosphorylated form of Chk1 was marginally increased in the top2–10 mutant cells after 2 h at a non-permissive temperature (Fig. 4(d)) with high percentage of aberrant mitosis at the same time point (Fig. 4(b)). This result further suggests that the aberrant chromatin structure generated at the time of mitosis could result in phosphorylation of Chk1 kinase protein although this activation of checkpoint response is not sufficient for the survival of top2–10 mutant cells.
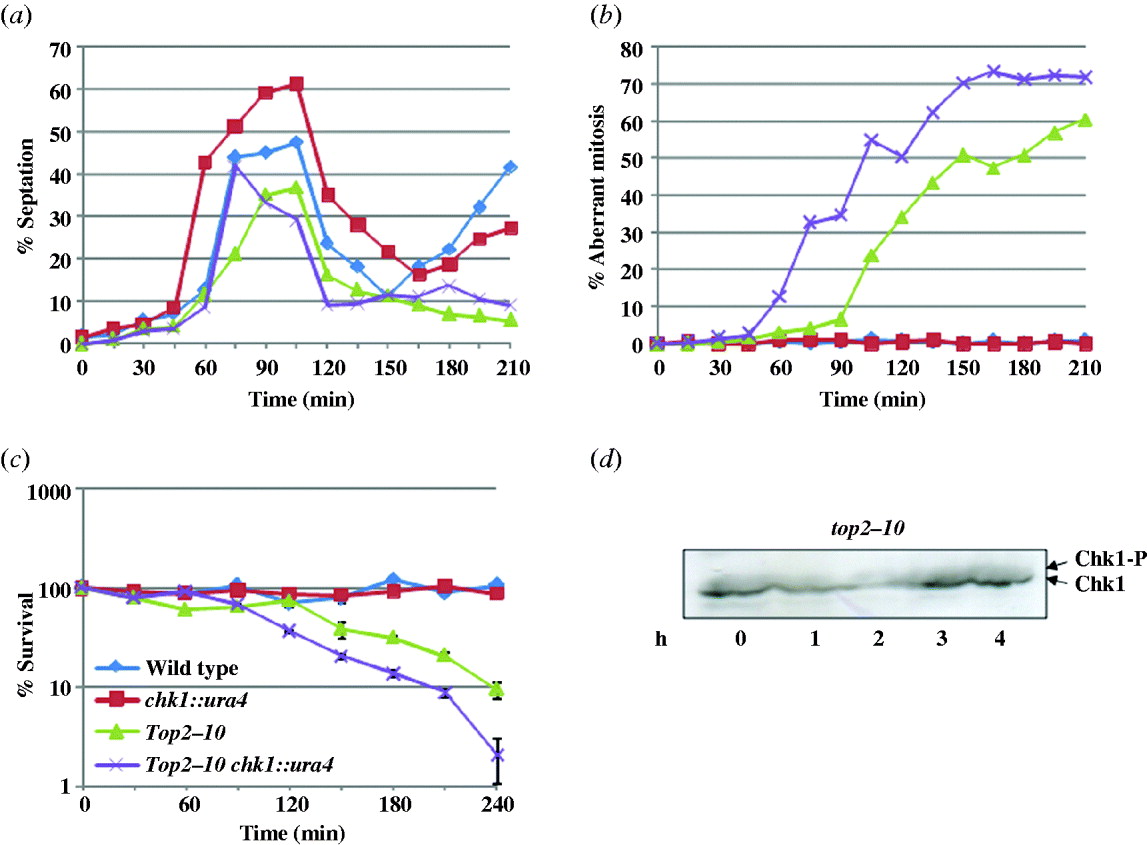
Fig. 4. Cells were synchronized in S phase and released at non-permissive temperature as described in section 2. Samples were collected at 15 min interval, fixed with 70% ethanol and stained with calcofluor or with DAPI. About 200 cells were counted for each sample, (a) percent septation and (b) percent aberrant mitosis was plotted. (c) Per cent survival in synchronized top2–10 mutant cells: indicated strains were processed as above, samples were collected at a 30 min interval; equal number of cells were plated on YEA plates and incubated at 25°C. The number of surviving colonies was calculated as the percentage of colonies appearing at permissive temperature. Values shown are the average of three independent experiments with standard deviation. (d) Chk1 phosphorylation in synchronized top2–10 mutant cells: cells were synchronized in S phase with 12 mM HU and released at non-permissive temperature as described in section 2. Samples were collected at 1 h time interval. Protein lysate was prepared and Western blot was performed using anti-HA antibody.
The absence of checkpoint function in the top2 mutant results in genome damage and genomic instability. A temperature-sensitive top2–10 mutant allele exhibits chromosome segregation defects. The double mutants of top2–10 and chk1 deletion lose their viability faster than the single top2–10 mutant at a non-permissive temperature with high percentage of aberrant mitosis as compared to single mutant suggesting that abrogating top2 function might be activating a checkpoint response involving Chk1. Since the top2–10 single-mutant cells lose their viability with the time suggest that the checkpoint response generated may not be sufficient for cell cycle delay. More recently, in Saccharomyces cerevisiae it has been shown that top1 and top2 localize at a chromosomal region undergoing replication and are required for solving the topological constraints at the moving replication fork and top2 mutant cells at a non-permissive temperature activate DNA damage checkpoint response (Bermejo et al., Reference Bermejo, Doksani, Capra, Katou, Tanaka, Shirahige and Foiani2007). It can be argued that due to the defective decatenating activity of top2 in fission yeast, topological constraints at the moving replication fork can be generated that can lead to DNA/chromatin damage at the time of mitosis and hence could activate a DNA damage checkpoint pathway. It is also possible that the aberrant chromosomal structure might be generated due to the entangled chromatid in top2–10 mutant cells at the time of mitosis and these defects are responsible for the activation of protein kinase Chk1. Furthermore, blocking the top2–10 mutant cells in G2 or exposing them with microtubule poison (TBZ) partially increases their survival at a non-permissive temperature suggest that loss in viability in top2–10 mutant cells could be due to the catastrophic defects, generated at the time of mitosis. In a previous study by Uemura & Yanagida (Reference Uemura and Yanagida1986), it has been shown that the viability of top2 mutant cells remained high during the G2 phase but decreased sharply at the time of mitosis. We speculate that the decrease in the viability in top2–10 mutant could be due to the catastrophic segregation of entangled chromatids. The aberrant chromosomal structure generated during mitosis in top2–10 mutant cells might generate signals that activate the protein kinase Chk1. Involvement of Chk1 pathway to delay metaphase to anaphase transition in the cells harbouring abnormal DNA structure has been shown in S. pombe and Drosophila (Collura et al., Reference Collura, Blaisonneau, Baldacci and Francesconi2005; Royou et al., Reference Royou, Macias and Sullivan2005). Further work is expected to unravel the mechanism in the top2 mutant in detail.
We are grateful to Dr Nancy Walworth for providing the strains. We thank Dr R. Ravishankar and Dr J. V. Pratap for critical reading of the manuscript and for helpful discussions. This work was supported by grants from Department of Biotechnology, India (GAP0027) and the Council of Scientific and Industrial Research (CSIR), India. The CDRI communication number for this manuscript is 7683.





