



Introduction
Wiedemann-Steiner syndrome (WSS, MIM #605130) is a Mendelian Disorder of Epigenetic Machinery (MDEM) most often caused by haploinsufficiency of KMT2A, a gene encoding a histone methyltransferase that adds histone 3 lysine 4 (H3K4) methylation (Jones et al., Reference Jones, Dafou, McEntagart, Woollard, Elmslie, Holder-Espinasse, Irving, Saggar, Smithson, Trembath, Deshpande and Simpson2012). In turn, disruption in chromatin remodeling results in secondary widespread abnormal gene expression and multisystemic developmental effects. This autosomal dominant disorder largely occurs de novo and is thought to affect males and females equally (Jones et al., Reference Jones, Dafou, McEntagart, Woollard, Elmslie, Holder-Espinasse, Irving, Saggar, Smithson, Trembath, Deshpande and Simpson2012).
WSS is extremely rare, which contributes to the challenges in characterizing the neurocognitive phenotype associated with this syndrome. While the first case of WSS was described in 1989 (Wiedemann et al., Reference Wiedemann, Kunze, Grosse, Dibbern, Wiedemann, Kunze, Grosse and Dibbern1989), it was not until 2012 that the genetic basis was discovered (Jones et al., Reference Jones, Dafou, McEntagart, Woollard, Elmslie, Holder-Espinasse, Irving, Saggar, Smithson, Trembath, Deshpande and Simpson2012). Hallmark characteristics of WSS includes hypertrichosis (arms, elbows and back), distinctive facial features (thick eyebrows, narrow palpebral fissures, wide spaced eyes, long eyelashes), and postnatal growth deficiency (Baer et al., Reference Baer, Afenjar, Smol, Piton, Gerard, Alembik, Bienvenu, Boursier, Boute, Colson, Cordier, Cormier-Daire, Delobel, Doco-Fenzy, Duban-Bedu, Fradin, Genevieve, Goldenberg, Grelet, Haye, Heron, Isidor, Keren, Lacombe, Lebre, Lesca, Masurel, Mathieu-Dramard, Nava, Pasquier, Petit, Philip, Piard, Rondeau, Saugier-Veber, Sukno, Thevenon, Van-Gils, Vincent-Delorme, Willems, Schaefer and Morin2018; Jones et al., Reference Jones, Dafou, McEntagart, Woollard, Elmslie, Holder-Espinasse, Irving, Saggar, Smithson, Trembath, Deshpande and Simpson2012; Miyake et al., Reference Miyake, Tsurusaki, Koshimizu, Okamoto, Kosho, Brown, Tan, Yap, Suzumara, Tanaka, Nagai, Nakashima, Saitsu, Niikawa and Matsumoto2016). Most notably, in a recent review of the largest clinical sample of patients with WSS to date, developmental delay and/or intellectual disability (mild to severe) was the most frequently observed feature among affected individuals, occurring in 97% of the clinical sample (Sheppard et al., Reference Sheppard, Campbell, Harr, Gold, Li, Bjornsson, Cohen, Fahrner, Fatemi, Harris, Nowak, Stevens, Grand, Au, Graham, Sanchez-Lara, Del Campo, Jones, Abdul-Rahman, Alkuraya, Bassetti, Bergstrom, Bhoj, Dugan, Kaplan, Derar, Gripp, Hauser, Micheil Innes, Keena, Kodra, Miller, Nelson, Nowaczyk, Rahbeeni, Ben-Schacher, Shieh, Slavotinek, Sobering, Abbott, Allain, Amlie-Wolf, Au, Bedoukian, Beek, Barry, Berg, Bernstein, Cytrynbaum, Chung, Donoghue, Dorrani, Eaton, Flores-Daboub, Dubbs, Felix, Fong, Fung, Gangaram, Goldstein, Greenberg, Ha, Hersch, Izumi, Kallish, Kravets, Kwok, Jobling, Knigh Johnson, Kushner, Lee, Levin, Lindstrom, Manickam, Mardach, McCormick, McLeod, Mentch, Minks, Muraresku, Nelson, Porazzi, Pichurin, Powell-Hamilton, Powis, Ritter, Rogers, Rohena, Ronspies, Schroeder, Stark, Starr, Stoler, Suwannarat, Velinov, Weksberg, Wilnai, Zadeh, Zand, Falk, Hakonarson, Zackai and Quintero-Rivera2021). However, to date, the specific neurocognitive profile associated with WSS remains elusive, as published literature largely involved retrospective review of medical charts (i.e., diagnoses made by a clinician) or natural history data.
Characterizing the neurocognitive and behavioral phenotypes associated with WSS is an important step towards elucidating the pathogenesis of cognitive disorders. Specifically, identifying particular cognitive functions impacted in this syndrome can offer clues on the downstream effect the disrupted epigenetic machinery has on neurodevelopment. For example, recent research involving Kabuki syndrome (KS), a disease caused by pathogenic variants in KMT2D, a gene with similar structure and function as KMT2A, was found to be associated with visuospatial deficits, suggestive of an aberration in the neurogenesis of the dentate gyrus (Harris et al., Reference Harris, Mahone and Bjornsson2019), which was consistent with memory impairment and histopathologic anomalies of the hippocampus in mouse models of this syndrome (Bjornsson et al., Reference Bjornsson, Benjamin, Zhang, Weissman, Gerber, Chen, Vaurio, Potter, Hansen and Dietz2014). In addition, the cognitive dysfunction and hippocampal neurogenesis deficits in mouse models of KS has been reported to be postnatally correctible with administration of a histone deacetylase inhibitor (Bjornsson et al., Reference Bjornsson, Benjamin, Zhang, Weissman, Gerber, Chen, Vaurio, Potter, Hansen and Dietz2014) or lysine-specific demethylase 1A inhibitor (Zhang et al., Reference Zhang, Pilarowski, Pich, Nakatani, Dunlop, Baba, Matsuda, Daini, Hattori, Matsumoto, Ito, Kimura and Bjornsson2021), and adopting a ketogenic diet (Benjamin et al., Reference Benjamin, Pilarowski, Carosso, Zhang, Huso, Goff, Vernon, Hansen and Bjornsson2017). Consequently, cognitive research involving individuals with KS and other disorders, such as that of Harris and colleagues (Reference Harris, Mahone and Bjornsson2019), is critical to inform therapeutic targets, further refining research done in animal models of the disease. Collectively, these findings in KS suggest that cognitive dysfunction associated with other MDEMs such as WSS may be reversible with appropriate treatment. Therefore, ascertaining the specific neuroanatomical and functional deficiencies linked to WSS will be central to the development of targeted clinical trials.
To this end, this case series aims to offer a more detailed description of the cognitive phenotype associated with WSS. Specifically, this investigation involves a retrospective chart review of neuropsychological assessment data in a clinical sample of patients with molecular confirmation of the genetic disorder. There is a suspicion of greater deficits in nonverbal and visuospatial skills, akin to the cognitive phenotype of KS (Harris et al., Reference Harris, Mahone and Bjornsson2019). This is particularly interesting given the structural similarities of the disease-relevant KMT2D (KS) and KMT2A (WSS) proteins involved in H3K4 histone methylation, which has a regulatory role in hippocampal functions in rodent models (Gupta et al., Reference Gupta, Kim, Artis, Molfese, Schumacher, Sweatt, Paylor and Lubin2010). Indeed, knockdown of KMT2A in mice similarly yielded memory impairment consistent with hippocampal dysfunction (Kerimoglu et al., Reference Kerimoglu, Sakib, Jain, Benito, Burkhardt, Capece, Kaurani, Halder, Agis-Balboa, Stilling, Urbanke, Kranz, Stewart and Fischer2017). Additionally, neuronal deletion of KMT2A in the prefrontal cortex, ventral striatum, and nucleus accumbens has been associated with working memory deficits and increased anxiety in mouse models, implicating that normal neurogenesis may rely on the downregulation of KMT2A (Jakovcevski et al., Reference Jakovcevski, Ruan, Shen, Dincer, Javidfar, Ma, Peter, Cheung, Mitchell, Jiang, Lin, Pothula, Stewart, Ernst, Yao and Akbarian2015; Shen et al., Reference Shen, Jiang, Javidfar, Kassim, Loh, Ma, Mitchell, Pothula, Stewart, Ernst, Yao, Martin, Shen, Jakovcevski and Akbarian2016). Accordingly, drawing from limited research with animal models as well as anecdotal experience in patients, we also anticipate deficits in executive functioning and emotion regulation.
Methods
Clinical sample
This case series involved a retrospective chart review of 10 pediatric patients (4F; M age at testing = 10.81 years, SD = 3.49; range: 6 to 17) with a molecularly confirmed diagnosis of WSS who were evaluated by the Department of Neuropsychology at Kennedy Krieger Institute (KKI) and followed by the Neurogenetics Clinic at KKI and/or the Epigenetics and Chromatin Clinic at Johns Hopkins University School of Medicine (JHUSOM). The racial composition of our sample was largely White (White, N = 9; Black, N = 1). All molecular testing was reviewed by a KKI or JHUSOM physician to confirm pathogenicity (whole exome sequencing, N = 9; single gene sequencing, N = 1) and all patients had at least one physical exam by those physicians to confirm the diagnosis of WSS. Majority of the patients had a pathogenic variant (N = 9), and one had a variant classified by the reporting laboratory as being of uncertain significance. However, we are confident that the latter variant is likely pathogenic and causing WSS in this individual because the patient’s findings are consistent with a diagnosis of WSS; it occurred de novo in the patient (i.e., it is not present in the unaffected parents); it has not been reported in large population databases like Genome Aggregation database (gnomAD), which report benign genetic variation; and a different amino acid substitution at the same position has been reported as pathogenic in an individual with features of WSS (Baer et al., Reference Baer, Afenjar, Smol, Piton, Gerard, Alembik, Bienvenu, Boursier, Boute, Colson, Cordier, Cormier-Daire, Delobel, Doco-Fenzy, Duban-Bedu, Fradin, Genevieve, Goldenberg, Grelet, Haye, Heron, Isidor, Keren, Lacombe, Lebre, Lesca, Masurel, Mathieu-Dramard, Nava, Pasquier, Petit, Philip, Piard, Rondeau, Saugier-Veber, Sukno, Thevenon, Van-Gils, Vincent-Delorme, Willems, Schaefer and Morin2018). Data of those with additional genetic anomalies such as microdeletions or chromosomal duplications were excluded to ensure the observed neurobehavioral patterns are specific to WSS. Our clinical sample included largely individuals with de novo variants in KMT2A (N = 7), one inherited from a parent with proven mosaicism, and two unknown inheritance. For more information on gene variant and pathogenicity stage, please see Supplementary Table 1.
Based on a review of the original assessment reports, all patients presented with functional speech at the time of the appointment. Additionally, vision and hearing were determined adequate for testing by clinicians. At the time, patient’s caregivers reported no significant concerns with vision or hearing. All patients were determined to show adequate effort, and test results were considered valid estimations of cognitive functioning based on the original clinician’s judgment. Out of our sample, eight patients had a diagnosis of intellectual disability and three with autism spectrum disorder.
This study was approved by the Institutional Review Board at JHU, and is in accordance with the Helsinki Declaration. Informed consent and/or assent were obtained by patient’s legal guardian and/or the patient prior to inclusion in the study.
Procedure and materials
All patients completed a comprehensive neuropsychological evaluation with a licensed psychologist at KKI as part of their clinical care. In total, four neuropsychologists completed testing with our patient sample and each provided a battery of cognitive measures based on their clinical judgment. Largely, as shown in Table 1, most patients completed a measure of intellectual functioning yielding nonverbal and verbal reasoning indexes, receptive vocabulary, visual/spatial perception, visuoconstruction, verbal learning/memory, visual memory, working memory, attention, and select academic skills (sight word reading, math calculation skills). Caregivers also completed a standardized parent-rating inventory to assess day-to-day executive functioning and adaptive skills.
Table 1. Proportion of patients (N = 10) that completed a test measure across cognitive domains

Abbreviations. Beery VMI-6 = Beery-Buktenica Developmental Test of Visual-Motor Integration 6th Edition, BRIEF = Behavior Rating Inventory of Executive Function, ChAMP = Child and Adolescent Memory Performance, DAS-II = Differential Ability Scales 2nd Edition, KTEA-3 = Kaufman Test of Educational Achievement 3rd Edition, NEPSY-II = A Developmental Neuropsychological Assessment, PPVT-5 = Peabody Picture Vocabulary Test 5th Edition, RIAS-2 = Reynolds Intellectual Assessment Scales 2nd Edition, WISC-V = Wechsler Intelligence Scale for Children 5th Edition, WIAT-III = Wechsler Individual Achievement Test 3rd Edition, WRAML-2 = Wide Range Assessment of Memory and Learning 2nd Edition.
Table 1 outlines the proportion of patients that completed each cognitive domain and tests administered. Half of the patients were evaluated via home-based teleneuropsychological testing with HIPAA-compliant videoconferencing platform (Zoom) during the COVID-19 pandemic. Those evaluated via telehealth modality were given a comparable test battery administered over one to two sessions, whereas patients seen face-to-face completed testing in a single session. The individuals who completed a teleneuropsychological assessment were administered subtests from the Wechsler Intelligence Scale for Children 5th Edition (WISC-V) and WISC-V Integrated to yield a WISC-V Verbal Comprehension Index and WISC-V Nonmotor Nonverbal Index with age-corrected norms (Raiford, Reference Raiford2017).
Supplementary table 2 lists the intellectual functioning composite for 8 of the 10 patients. This estimate was not obtained for two patients as one completed only verbal and nonverbal cluster subtests from the Differential Ability Scales 2nd Edition (DAS-II) School-Age battery and the other completed the out-of-level DAS-II Early Years battery. Both assessments did not yield the DAS-II General Conceptual Ability composite, an index of intellectual functioning. Of the eight patients, four completed WISC-V and WISC-V Integrated subtests through telehealth modality which yielded the Nonmotor General Ability Index (Nonmotor GAI)(Raiford, Reference Raiford2017); two completed WISC-V subtests which were used to generate the GAI, one received an intelligence index through completion of the Reynolds Intellectual Assessment Scales 2nd Edition (RIAS-2), and one had the General Conceptual Ability composite through the DAS-II.
Data strategy
Across standardized cognitive and academic test measures, the derived performance scores were converted to standard scores and percentile ranks utilizing age-corrected norms in the respective technical and scoring manuals. Given our small sample, nonparametric testing was used. Wilcoxon signed-rank test was applied to determine if a within-group difference in verbal and nonverbal indexes was observed. Finally, ratings provided for the Behavior Rating Inventory of Executive Function (BRIEF) and Behavior Rating Inventory of Executive Function 2nd Edition (BRIEF-2) were converted based on gender- and age-based norms. Subsequently, patients’ objective test performance across each cognitive domain was qualitatively coded based on the resulting percentile rank: Low to Very Low for ≤ 2nd percentile, Below Average for performance between 3rd and 15th percentile, Within Normal Limits or Above when ≥16th percentile. Likewise, resulting standard scores on caregiver inventories were coded for clinical significance or marked functional impairment. On the BRIEF/BRIEF-2, in accordance to the inventory manual, Clinically Significant refers to obtained T-scores ≥70, At-Risk (i.e., Elevated to Potentially Clinically Significant) refers to T-scores 60 to 69, and Within Normal Limits reflects T-scores ≤59.
Results
Cognitive test performance
As shown in Table 2, on average, patients with WSS yield stronger performance on verbal/language measures than nonverbal and visual processing tests. Average percentile rank for Receptive Vocabulary and Verbal Memory were within normal limits or above; whereas, mean performance on Nonverbal Index (nonverbal reasoning), Visual and Spatial Perception, Visuoconstruction, and Visual Memory were well below average. Verbal reasoning was slightly below average. Wilcoxon signed-rank test revealed Nonverbal Index scores were significantly lower than Verbal Index scores, Z = -2.89, p = .005. However, verbal and language measures also yielded more variable performance as reflected by wider range in percentile ranks, as compared to nonverbal and visual perceptual tests. On average, Attention and Working Memory were below average, although the latter also yielded a wide range of outcome scores. Among academic measures, patients with WSS showed relatively stronger sight word reading with performance scores that fell between below average to average, as compared to math computation skills, which was very low to below average.
Table 2. Average percentile rank across cognitive domain
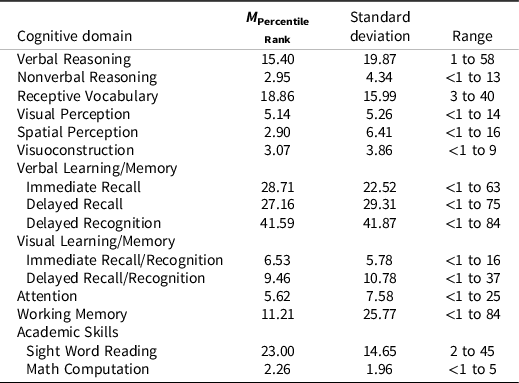
Note. Percentile rank of 16th and above is considered within normal limits or above, 3rd to 15th as below average, and ≤2nd as low to very low.
Supplementary table 2 outlines individual patient’s percentile rank across cognitive domains and their intellectual functioning composite (standard score). As illustrated in the table, the pattern of stronger verbal over nonverbal reasoning skills was observed regardless of testing modality. Specifically, four of the five patients who completed telehealth testing and three of five patients who completed face-to-face or an in-person neuropsychological evaluation showed stronger Verbal than Nonverbal Index scores.
Figure 1 reflects the proportion of patients who performed within normal limits or above across cognitive domains. In brief, approximately 30% patients performed within normal limits or above in Verbal Index (3 out of 10), 57.14% in Receptive Vocabulary (4 out of 7), 77.77% in Verbal Immediate Memory Recall (7 out of 9), 44.44% in Verbal Delayed Memory Recall (4 out of 9), 55.55% in Verbal Delayed Recognition (5 out of 9), and 75% in Sight Word Reading (6 out of 8). One patient or 10% of the clinical sample performed within normal limits in Attention and Working Memory, 11.11% in Visual Immediate and Delayed Memory (1 out of 9), and 16.67% in Spatial Perception (1 out of 6). In contrast, no patients performed within broad average or above in Nonverbal Index, Visual Perception, Visuoconstruction, or Math Computation.
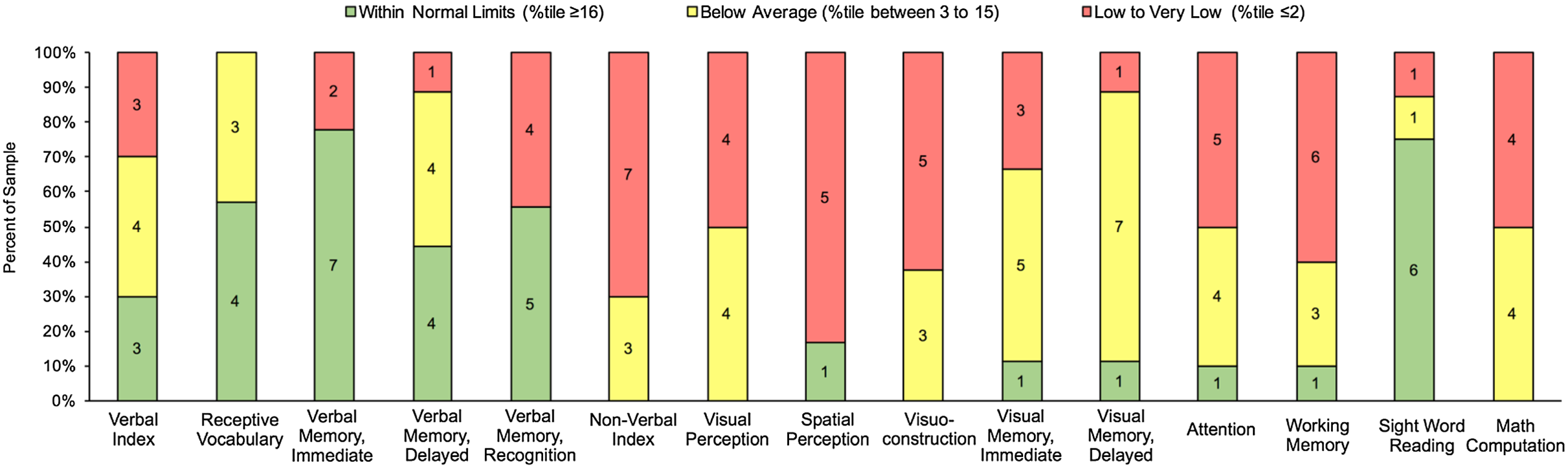
Figure 1. Number of patients performing within normal limits or above, below average, and low to very low across cognitive domains.
Parent report inventory of executive functioning
Figure 2 illustrates the percentage of the clinical sample whose parents’ ratings revealed at-risk to clinically significant problems across executive functioning domains. Of note, one patient completed the BRIEF, which does not yield an Emotion Regulation Index. Overall, the majority of the clinical sample (90%) were rated in the at-risk or clinically significant range for global problems with executive functioning. Across executive domains, most concerns were reported in Emotion Regulation (100% or 9 out of 9), followed by Behavior Regulation (80% or 8 out of 10) and Cognitive Regulation (70% or 7 out of 10). Of note, Pearson bivariate correlations did not reveal associations between intellectual functioning composite and executive domains or the overall BRIEF General Executive Composite.

Figure 2. Parent ratings on the Behavior Rating Inventory of Executive Functioning (BRIEF or BRIEF-2).
Discussion
This case series is the first study to provide a broad overview of the neurocognitive profile associated with pediatric patients with confirmed pathogenic variants in KMT2A. In brief, on average, those with WSS showed relatively stronger verbal reasoning, verbal memory and receptive language than nonverbal reasoning, visual memory, and visual-spatial processing. Sight word reading was also observed as an area of relative strength, whereas math computation was a consistent area of academic difficulty. The majority of patients showed below average to very low performances across attention and working memory; and were rated in the at-risk to clinically significant range in day-to-day executive functioning deficits.
Collectively, our clinical sample of pediatric patients with WSS revealed more consistent pattern of impairment in nonverbal reasoning, visual/spatial perceptual, and object learning/memory. While verbal and select language skills appear relatively more intact, greater heterogeneity was observed among these functions. Broadly, our findings may suggest hippocampal dysfunction, particularly given observed deficiencies in visual memory, visuospatial perception and visual integration in both motor and mental construction tasks (Burgess, Reference Burgess2008; Lee et al., Reference Lee, Yeung and Barense2012; Wicking et al., Reference Wicking, Nees and Steiger2014). These observed cognitive trends are congruent with the phenotypic features of individuals with KS (Harris et al., Reference Harris, Mahone and Bjornsson2019). As highlighted by Harris and colleagues (Reference Harris, Mahone and Bjornsson2019), these pattern of findings may implicate the effect of the gene mutation on hippocampal, and specifically dentate gyrus, neurogenesis following disruption in methylation of H3K4 and subsequent chromatin remodeling. Notably, other studies involving animal models have observed deficits in hippocampal functions, including memory formation, with deficiency in H3K4 histone methyltransferase (Gupta et al., Reference Gupta, Kim, Artis, Molfese, Schumacher, Sweatt, Paylor and Lubin2010). However, given the limited cognitive constructs assessed and the mixed tests administered, additional research with larger sample sizes and comparison groups, supplemental metrics of neural functioning (e.g., electroencephalogram, EEG; positron emission tomography, PET), and a comprehensive battery of measures directed towards the examination of hippocampal functions are necessary to ascertain if our observed pattern of cognitive functioning reflects hippocampal dysfunction uniquely or byproducts of other neurodevelopmental anomalies. While neurocognitive testing offers critical clues on brain function that may not be reflected on structural or anatomical imaging studies (e.g., MRI), cognitive functions generally involve multiple neural substrates/circuits. As such, neuropsychological assessment should not be utilized alone to isolate functional deficits to a single brain region. Integration of EEG, PET or fMRI studies with cognitive and behavioral testing will be imperative in subsequent investigations to identify focal brain dysfunction associated with WSS.
Importantly, future cross-syndrome comparisons of the cognitive and behavioral phenotypes associated with mutations in the KMT2 family of genes (e.g., KS with pathogenic variant of KMT2D, intellectual disability or autism related to KMT2C mutation)(Vallianatos & Iwase, Reference Vallianatos and Iwase2015) and Mendelian Disorders of the Epigenetic Machinery broadly (Fahrner & Bjornsson, Reference Fahrner and Bjornsson2019; Mossink et al., Reference Mossink, Negwer, Schubert and Nadif Kasri2021) will better elucidate the shared pathogenic mechanism that results in ontogenesis of developmental disorders and psychopathology, and likewise, unique disease characteristics. These efforts will better inform targets of syndrome-specific clinical trials, and likewise epigenetic therapies that can be generalized across syndromes with common disease-causing mechanisms.
Impaired working memory and executive functioning based on objective and parent-report measures may also implicate the role of KMT2A in the neurogenesis of the prefrontal cortex (Jakovcevski et al., Reference Jakovcevski, Ruan, Shen, Dincer, Javidfar, Ma, Peter, Cheung, Mitchell, Jiang, Lin, Pothula, Stewart, Ernst, Yao and Akbarian2015). Executive dysfunction, including poor emotion and behavior regulation, may explain the relatively high rate of hyperactivity (44.3%), and aggressive behaviors (33%) that has recently been documented with a large cohort of patients with WSS (Sheppard et al., Reference Sheppard, Campbell, Harr, Gold, Li, Bjornsson, Cohen, Fahrner, Fatemi, Harris, Nowak, Stevens, Grand, Au, Graham, Sanchez-Lara, Del Campo, Jones, Abdul-Rahman, Alkuraya, Bassetti, Bergstrom, Bhoj, Dugan, Kaplan, Derar, Gripp, Hauser, Micheil Innes, Keena, Kodra, Miller, Nelson, Nowaczyk, Rahbeeni, Ben-Schacher, Shieh, Slavotinek, Sobering, Abbott, Allain, Amlie-Wolf, Au, Bedoukian, Beek, Barry, Berg, Bernstein, Cytrynbaum, Chung, Donoghue, Dorrani, Eaton, Flores-Daboub, Dubbs, Felix, Fong, Fung, Gangaram, Goldstein, Greenberg, Ha, Hersch, Izumi, Kallish, Kravets, Kwok, Jobling, Knigh Johnson, Kushner, Lee, Levin, Lindstrom, Manickam, Mardach, McCormick, McLeod, Mentch, Minks, Muraresku, Nelson, Porazzi, Pichurin, Powell-Hamilton, Powis, Ritter, Rogers, Rohena, Ronspies, Schroeder, Stark, Starr, Stoler, Suwannarat, Velinov, Weksberg, Wilnai, Zadeh, Zand, Falk, Hakonarson, Zackai and Quintero-Rivera2021), in addition to case reports of autism-like features including rigid and externalizing behaviors but intact social motivation (Chan et al., Reference Chan, Cytrynbaum, Hoang, Ambrozewicz, Weksberg, Drmic, Ritzema, Schachar, Walker, Uddin, Zarrei, Yuen and Scherer2019). Expanding the clinical phenotypic spectrum of WSS by utilizing a developmental approach will be a vital step to discern developmental psychopathology that emerges as downstream effects of the disrupted epigenetic machineries. Moreover, longitudinal approaches in phenotyping efforts are necessary to determine the stability of the neurobehavioral profile, and/or sensitive periods when clinical and behavior interventions may offer more protective effects on long-term cognitive sequelae.
There are limitations in our case series that prospective research should consider. Given the rarity of WSS, our clinical sample is small. A larger sample will be necessary to illustrate precise epigenotype-phenotype associations. Given neuropsychological evaluations were administered to patients for clinical referrals, heterogeneous tests with different task instructions were combined to denote a cognitive domain (e.g., NEPSY-II Arrows offers increasing number of possible response options in later items, whereas JLO has a consistent number of choices). In addition, the assessment batteries provided included limited measures of hippocampal functioning. Although neurocognitive measures are sensitive to functionality of neural substrates (Wicking et al., Reference Wicking, Nees and Steiger2014), these often index a combination of cognitive skills and thus multiple neural systems involved. Thus, future studies should consider providing a targeted battery of tests that assess spatial cognition and memory in addition to episodic memory – all of which are dedicated functions of the hippocampus. Offering a wider range of cognitive assays that collectively implicate various aspects of the hippocampal formation will afford more precise measurements and strengthen the gene-brain-cognitive associations driven by the epigenetic imbalance resulting from KMT2A mutation. The heterogeneity of our clinical sample (i.e., wide age range of patients) should also be considered, as cognitive and academic constructs may develop at different pace across select developmental periods. Notably, impairment in select cognitive functions may be associated with differential impact on day-to-day functioning based on the developmental stage. Finally, our clinical sample included a combination of patients evaluated through telehealth and face-to-face modalities. The proportion of those with impairment under the telehealth versus face-to-face in-clinic modality were roughly similar across Verbal Index (2/5 vs. 1/5) and Nonverbal Index (3/5 vs. 4/5). While there are limitations to teleneuropsychology (e.g., there are more limited measures adapted for remote virtual administration than traditional face-to-face testing), the swift adoption of this model of care secondary to the pandemic has revealed its utility in increasing access to clinical services and research for some families affected by rare diseases such as WSS. Nevertheless, more research is needed to support the validity of in-home teleneuropsychological testing particularly among pediatric samples with greater cognitive and physical/sensory impairment, which are typically more prevalent in syndromic intellectual disability.
In brief, our case series with a sample of pediatric patients with WSS observed a distinctive trend of discrepant impairment in nonverbal reasoning, visual-spatial perception, visual memory and math calculation skills that warrant more investigation. Systematic and longitudinal approach to cognitive phenotyping will be necessary to identify if these deficits vary as a function of variant or other molecular characteristics.
Supplementary material
To view supplementary material for this article, please visit https://doi.org/10.1017/S1355617722000467
Acknowledgements
We would like to thank the patients and their families who participated in this study, as well as acknowledging the support of the Wiedemann-Steiner Syndrome Foundation.
Funding statement
No specific funding was received for this study. However, R.N. would like to acknowledge support from National Institute of Health (NIH)(R25 NS117356). J.H. has support from the National Institute of Child Health and Development (NICHD)(K23HD101646), J.A.F. from The Hartwell Foundation (Individual Biomedical Research Award) and the NIH/NICHD (K08HD086250), and H.T.B. from the Wiedemann-Steiner Syndrome Foundation, Louma G. Foundation, Icelandic Research Fund (195835, 206806, 217988) and Icelandic Technological Development Fund (2010588).
Conflicts of interest
H.T.B. is a consultant for Mahzi therapeutics.







 Open access
Open access