


SCFA such as butyrate, propionate and acetate are the endproducts of dietary carbohydrate breakdown by anaerobic bacteria in the large bowel. Butyrate is utilised by colonocytes for energetic purposes.
Topical therapy with butyrate appears to be a promising development in the treatment of inflammatory bowel disease. The molecular mechanisms leading to the response to SCFA, and especially towards butyrate, are poorly understood. The mechanisms do not seem to be limited to the view that butyrate only supplies the energy for colonocytes. Recent studies have demonstrated that butyrate exerts immunomodulatory effects, such as down regulation of T-cell responses, induction of Th1 cell anergy and modulation of antigen-presentation-associated molecules (Diakos et al. Reference Diakos, Prieschl, Saemann, Novotny, Bohmig, Csonga, Baumruker and Zlabinger2002). Additionally, a recent study has demonstrated that butyrate decreases pro-inflammatory cytokine expression via inhibition of NFκB activation and inhibitory κBα degradation (Luhrs et al. Reference Luhrs, Gerke, Muller, Melcher, Schauber, Boxberge, Scheppach and Menzel2002). Less consideration has been given to the possible use of butyrate-producing bacteria as probiotics. A number of studies focusing on culturable human intestinal microflora (Frankel et al. Reference Frankel, Zhang, Singh, Klurfeld, Don, Sakata, Modlin and Rombeau1994; Wilson et al. Reference Wilson, Hall, Brazier and Lewis2000; Duncan et al. Reference Duncan, Barcenilla, Stewart, Pryde and Flint2002, Reference Duncan, Scott, Ramsay, Harmsen, Welling, Stewart and Flint2003, Reference Duncan, Holtrop, Lobley, Calder, Stewart and Flint2004a , Reference Duncan, Louis and Flint b ; Pereira & Gibson, Reference Pereira and Gibson2002; Vine et al. Reference Vine, Charman, Gibson, Sinclair and Porter2002; Louis et al. Reference Louis, Duncan, McCrae, Millar, Jackson and Flint2004) have demonstrated that the colon harbours significant populations of butyrate-producing bacteria, such as Clostridium, Eubacterium, Fusobacterium, etc. Recent studies have concentrated on combining molecular and cultural approaches to investigate the dominant butyrate-producing bacteria of the human colon and have identified a large number of butyrate-producing isolates based on sequencing and restriction fragment length polymorphism analyses of 16S rRNA genes.
A probiotic is considered a live microbial feed supplement that has beneficial effects on the host by improving its intestinal microbial balance (O'Sullivan et al. Reference O'Sullivan, Kelly, O'Halloran, Collins, Collins, Dunne and Shanahan2005) or ‘a live microbial food ingredient that is beneficial to health’ (Otles et al. Reference Otles, Cagindi and Akcicek2003). The criteria for a micro-organism to be defined as probiotic include that the strain be of human origin, be safe for human use, be stable in acid and bile acids, and adhere to the intestinal mucosa (Chen & Walker, Reference Chen and Walker2005). Probiotic strains are often selected on an empirical basis from bacterial pools and include human and traditional fermented food isolates. Probiotic bacteria are shown to promote the host's endogenous defence mechanisms by enhancing the humoral immune responses and thus promoting the intestine's immunological barrier (Saggioro, Reference Saggioro2004). Moreover, probiotic bacteria have been shown to stimulate non-specific host resistance to microbial pathogens (Resta-Lenert & Barrett, Reference Resta-Lenert and Barrett2003), and thereby aid in immune elimination, and modulate the host's immune responses to potentially harmful antigens with the potential to down regulate hypersensitivity reactions (Snelling, Reference Snelling2005). A previously reported study (Djouzi et al. Reference Djouzi, Andrieux, Degivry, Bouley and Szylit1997) demonstrated an increase in SCFA in the caeca of germ-free rats inoculated with human intestinal microflora and fed fermented milks without a concomitant change in lactate concentration. The authors concluded that the increase in SCFA was beneficial for the host since they were absorbed from the colon and provided an important source of energy for tissues, especially of butyrate, which is the preferred energy source for colonocytes.
The aim of the present study was to isolate and characterise in-depth a putatively functional butyrate-producing facultative anaerobe, a bacterium belonging to the species Enterococcus durans. We tested the ability of this novel bacterial isolate to suppress inflammation and concomitantly ameliorate the symptoms of dextran sodium sulfate (DSS)-induced colitis in mice.
Materials and methods
Faecal sample preparation
The bacterium described in the present paper was isolated from the fresh faeces of a healthy human vegetarian. The volunteer had not consumed any antibiotics or other drug known to affect faecal flora for at least 6 months before taking the faecal sample. Sterile peptone solution (9 ml; 0·1 %, w/v) in water was added to 1 g faeces and then mixed with a stomacher. A dilute faecal sample was incubated in liquid intestinal microflora medium (IMM) containing 15 ml rumen fluid, 0·2 g peptone, 0·2 g bacto soytone, 0·1 g yeast extract, 1 × 10− 4 g resazurin, 0·4 g Na2CO3, 0·05 g cysteine hydrochloride, 0·5 g carboxymethylcellulose, 5 ml of a mineral solution containing (per litre): 0·004 g KH2PO4, 0·09 g NaCl, 0·009 g MgSO4.7H2O, 0·009 g CaCl2.2H2O, 0·014 g (NH4)2SO4, 15 × 10− 6 g MnSO4.4H2O, 18 × 10− 7 g CoCl2.6H2O, 0·5 ml of a vitamin solution containing per litre: 0·1 mg B12, 20 mg thiamine hydrochloride, 20 mg Ca-d-pantothenate, 20 mg pyridoxine hydrochloride, 20 mg nicotinamide, 20 mg riboflavin, 1 mg p-aminobenzoic acid, 0·25 mg biotin and 0·25 mg folic acid, essentially as previously described by Barcenilla et al. (Reference Barcenilla, Pryde, Martin, Duncan, Stewart, Henderson and Flint2000). The dilute faecal sample was incubated in IMM broth under microanaerobic conditions (using anaerobic sachets AN-35 (Oxoid Unipath Ltd, Basingstoke, Hants, UK) in anaerobic chambers) at 37°C, and at different pH: 5·0, 5·5, 6·0, 6·5, 7·0, 7·5, 8·0 and 9·6.
Quantitative detection of butyrate
Detection and quantification of butyrate concentration was performed following injection of a sample of growth medium into a gas chromatograph with a flame ionisation detector and capillary column (HP-FFAP 30 m × 0·53 mm × 1·0 μm). Running conditions were as described in the capillary column instructions (Agilent Technologies, Palo Alto, CA, USA). Briefly, the samples were injected at 240°C, He was run at 15 ml/min and the flame ionisation detector was brought to 240°C. The initial temperature was 40°C for 2 min. and then rose gradually at 30°C/min or 35°C/min, until the desired temperature was reached.
Selection of high-butyrate-producing bacterial isolates
The faecal sample bacteria were inoculated on IMM agar for 48 h and fifty colonies were then randomly chosen. The colonies were incubated in IMM broth and those which secreted the highest quantities of butyrate were selected for further characterisation.
Identification of the faecal isolate producing butyrate
Gram staining was performed as described in the Gram stain kit instructions (Difco Laboratories, Detroit, MI, USA).
The selected bacterial isolate was grown in 10 ml De Man–Rogosa–Sharpe (MRS) medium (Difco Laboratories) for one night. Putative bacterial plasmids were isolated using the Qiagen Plasmid Mini purification kit (Hilden, Germany) according to the instruction manual. Isolated and purified plasmids were monitored on an agarose-analytical gel.
Genomic DNA was isolated from the bacterial isolate by the genomic DNA purification kit (Promega, Madison, WI, USA). The nucleotide sequence of the reverse primer used in the PCR was 1392r universal 5′-ACGGGCGGTGTGTAC-3′ and the nucleotide sequence of the forward sequence was 11f universal 5′-GTTTGATCCTGGCTCAG-3′ (Sigma Chemicals, St Louis, MO, USA). PCR amplification was performed by a PCR thermal cycler under the following conditions: denaturation at 95°C for 1 min, followed by twenty-nine cycles of denaturation at 94°C for 30 s, annealing at 44°C for 40 s, and extension at 72°C for 1 min and 40 s. After completion, an additional extension step was performed at 72°C for 4 min, and the samples were then chilled to 4°C. The final mixture volume was 50 μl, and contained deoxynucleotide triphosphates (2 mmol/l), 4 mm-MgCl2, bovine serum albumin (250 μg/ml), 30 μl PCR water, forward primer (50 pmol/μl), reverse primer (50 pmol/μl), 1·5 μl red Taq DNA polymerase and its 5 μl buffer (Sigma) and purified bacterial genomic DNA (30 ng/μl). The primers were tested in PCR amplification with E. coli genomic DNA. Analyses were performed by Blast on the website of the National Center of Biotechnology Information (www.ncbi.nlm.nih.gov/blast/Blast.cgi).
Butyrate kinase sequence analysis
The nucleotide sequences of the PCR primers were chosen from the genomic DNA sequence of the butyrate-producing bacterium Clostridium acetobutylicum taken from the website of the National Center of Biotechnology Information. The nucleotide sequence of the forward primer used in PCR was 5′-TGCGTCAAATCTTGGTGGAA-3′, which is located at 1385–1404; that of the reverse sequence was 5′-AAGTACCTCCACCCAGGTGT-3′, which is located at 1615–1635 (MWG-Biotech AG, Reinach, Switzerland). PCR amplification was performed by a gradient-PCR thermal cycler under the following conditions: denaturation at 95°C for 5 min, followed by thirty-five cycles of denaturation at 95°C for 30 s, annealing at 40°C to 60°C (with 2°C jumps) for 1 min, and extension at 72°C for 1 min. After completion, an additional extension step was performed at 72°C for 5 min, and the samples were then chilled to 4°C. The final mixture volume was 50 μl, and contained deoxynucleotide triphosphates (10 mmol/l), MgSO4 (25 mmol/l), 31 μl PCR water, forward primer (10 mmol/l), reverse primer (10 mmol/l), 1·5 μl Tfl Taq DNA polymerase and its 10 μl buffer (RT PCR kit; Promega) and purified bacterial genomic DNA (25 ng/μl).
All PCR products were visualised in ethidium bromide-stained agarose gels, purified by gel extraction kit (Qiagen; Westburg, Leusden, The Netherlands) and nucleotide sequences were determined by automated DNA sequencer (ABI 3100) at the DNA analysis laboratory, Faculty of Science of the Hebrew University of Jerusalem (Israel).
Gelatin zymography
We examined the bacterial isolate's ability to synthesise some of the harmful factors previously described to be produced by pathogenic Enterococcus strains, such as gelatinases. To this end zymographic analyses using gelatin gels were performed on concentrated medium samples from bacterial cultures essentially as described by Kawalec et al. (Reference Kawalec, Potempa, Moon, Travis and Murray2005) with modifications. Samples were treated with Laemmli SDS-PAGE sample buffer for 30 min at 37°C, subjected to electrophoretic separation on a gelatin-impregnated (1 mg/ml; Difco) SDS-8 % polyacrylamide gel under non-reducing conditions, followed by 30 min of shaking in 2·5 % Triton X-100 (BDH, Poole, Dorset, UK). The gels were then incubated for 16 h at 37°C in tri(hydroxymethyl)-aminomethane (50 mmol/l), NaCl (0·2 mol/l), CaCl2 (5 mmol/l) at pH 7·6. At the end of the incubation, the gels were stained with 0·5 % (w/v) Coomassie Blue G 250 (Bio-Rad, Richmond, CA, USA) in methanol–acetic acid–water (30:10:60, by vol.).
Effects of low pH and bile acids on growth rate of selected bacterial isolate
Selected bacterial colonies were grown to mid log phase (up to 0·4–0·5 optical density units at 600 nm) and then incubated for 1, 2, 3 and 4 h at 37°C in MRS medium, adjusted to pH 2 or 3 with HCl (5 mol/l). The number of surviving bacteria was determined by a plate-count procedure on MRS agar at pH 6·0. The sensitivity of the selected bacterial colonies to bile was tested by incubating the bacteria in MRS medium containing 0·05 % (w/v) ox-gall (Oxoid Unipath Ltd) for 2, 4 and 6 h. MRS medium at pH 6·0 was used as a control. The number of surviving bacteria was again determined by the plate-count procedure as above. The number of colonies was counted after each incubation time. All the experiments were performed in quadruplicate.
E. durans was grown in IMM as described previously. MRS medium was used to maintain the Enterococcus isolate. Lactobacillus delbrueckii subsp. lactis ATCC 4797 was grown in MRS broth at 37°C without shaking. Bacterial isolates were stored in appropriate media containing 50 % (v/v) glycerol at − 80°C.
Assays in tissue cultures
A Caco-2 cell line, originally isolated from a human colon adenocarcinoma, was obtained from ATCC (number: HTB-38). Cells were routinely grown at 37°C in a 5 % CO2–95 % air atmosphere in Dulbecco's modified Eagle's medium with 1 % (v/v) l-glutamine, 10 % (v/v) fetal calf serum and 0·2 % (v/v) penicillin (Biological Industries, Kibbutz Beit Haemek, Israel). Cells were seeded at a concentration of 105 cells/ml. The culture medium was replaced every 2 d.
In vitro adhesion assay
Caco-2 monolayers at late stages of post-confluence (after 14 d of culture) were prepared on plastic coverslips, which were placed in eight-well tissue culture plates. The selected E. durans was compared with additional established probiotic bacteria, L. rhamnosus GG and L. casei Shirota, which were included as high- and low-adhesive strains (Tuomola & Salminen, Reference Tuomola and Salminen1998), respectively. Overnight bacterial cultures (50 μl) in 5 ml of MRS medium and 200 μl Dulbecco's modified Eagle's medium without additions were added to each well of the tissue culture plate and incubated. After 0·5, 1, 1·5 and 2 h of incubation, the monolayers were washed twice with sterile PBS, fixed with 95 % (v/v) methanol, stained with Giemsa stain solution (1:20) (Sigma), washed until no colour was observed in the washing solution, dried and examined microscopically at 1000 × magnification under oil immersion. For each monolayer on a coverslip well, the number of cells with adherent bacteria was evaluated compared with the number of cells without adherent bacteria in ten random microscopic areas. This comparison represents the adherence percentage. The adhesion capacity was determined by counting Giemsa-stained Caco-2 and bacterial cells under a microscope at 1000 × magnification.
Effect of Enterococcus durans on dextran sodium sulfate-induced colitis
The experiments were conducted in full compliance with the strict guidelines of the Hebrew University policy on animal care and use. Colitis was induced in male 10- to 12-week-old Balb/c mice (Harlan Laboratories Ltd, Rehovot, Israel), which were randomly divided into three different groups of six mice each. The DSS group received 2·5 % (w/v) DSS (Sigma) in the drinking water and was fed only Purina chow (Harlan Laboratories Ltd, Rehovot, Israel), whereas the control group received tap water and Purina chow. There were two experimental groups: one received 5 % (w/v) DSS in the drinking water and fed Purina chow containing 1 × 108 colony-forming units (cfu) L. delbrueckii/g; the other also received 5 % DSS in the drinking water as above and fed Purina chow containing 1 × 108 cfu E. durans/g.
We choose as control the food-grade lactic acid bacterium L. delbrueckii subsp. lactis ATCC 4797, since its measured ability to produce butyrate as assessed by gas chromatograph methodology resulted in negligible concentrations as compared with E. durans (data not shown).
We evaluated animal body weight, stool consistency and the presence of blood in the stools daily. After 8 d of DSS treatment and feeding with the different diets, mice were killed and tissue samples were harvested from the colon. The tissue samples were removed and divided. One portion was snap-frozen in liquid N2 for evaluation of myeloperoxidase (MPO) activity. Another was fixed in buffered formalin (4 %, v/v) at 4°C and embedded in paraffin, and then sections (5 μm thick) stained with haematoxylin and eosin were subjected to histological evaluation or fluorescent in situ hybridisation (FISH) analysis to assess the presence of E. durans adhered to the colonic mucosa.
The extent of clinical activity in DSS-induced colitis was determined and scored according to typical clinical signs of intestinal inflammation (disease activity index; DAI). Scores were defined as follows: loss in body weight, 0 = no loss; 1 = 5 to 10 %; 2 = 11 to 15 %; 3 = 16 to 20 %; 4 = over 20 %; haemoccult, 0 = no blood; 2–3 = positive; 4 = gross blood.
Histological findings were also graded according to stage of intestinal inflammation, using a previously described grading scale (Schultz et al. Reference Schultz, Strauch, Linde, Watzl, Obermeier, Gottl, Dunger, Grunwald, Scholmerich and Rath2004) with the following criteria: no inflammation = 0; slightly disturbed mucosal architecture = 1; more disturbed mucosal architecture = 2; marked mucosal hyperplasia = 3; severe hyperplasia and mucosal ulcerations = 4.
Determination of myeloperoxidase activity
Colonic tissue samples (about 100–120 mg) were homogenised in 50 mm-potassium phosphate buffer (pH 6·0, Sigma) and centrifuged at 41 400 g for 10 min; pellets were suspended in 50 mm-potassium phosphate buffer containing 0·5 % (w/v) hexadecyltrimethylammonium bromide (Sigma). After three freeze-and-thaw cycles, with sonication between cycles, the samples were centrifuged at 41 400 g for 10 min. Samples (0·3 ml) were added to 2·3 ml of reaction mixture containing 50 mm-potassium phosphate buffer, o-dianisidine (Sigma), and H2O2 solution (20 mmol/l). One unit of enzyme activity was defined as the amount of the MPO present that caused a change in absorbance measured at 460 nm for 3 min. MPO activity was expressed as U/g tissue.
Expression analyses of colonic interleukin-1β and tumour necrosis factor-α
Total RNA was isolated from colonic tissue samples homogenised in Tri Reagent solution (MRC, Cincinnati, OH, USA). RT-PCR assay was performed using the Promega kit assay. The selected primers were as follows: IL-1β (sense) 5′-ATGAAGCTTTGTACAAGGAGAACCA-3′; (antisense) 5′-TTAGGAAGACACAGATTCCATGGT-3′; TNF-α (sense) 5′-ATCAGTTCTATGGCCCAGACCT-3′; (antisense) 5′-TCACAGAGCAATGACTCCAAAGTA-3′. Glyceraldehyde-3-phosphate dehydrogenase primers were used for RNA control loading: (sense) 5′-ACCACAGTCCATGCCATCAC-3′; (antisense) 5′-TCCACCACCCTGTTGCTGTA-3′. The PCR protocol was 5 min at 94°C, then twenty-eight cycles (1 min, 94°C, 1 min, 68°C, 1 min, 72°C) for the test genes and twenty cycles at similar conditions for glyceraldehyde-3-phosphate dehydrogenase, and a final 10 min at 72°C for all samples.
Localisation of butyrate-producing bacteria by fluorescent in situ hybridisation analyses
The FISH method was used to determine whether the bacterial isolate adhered to the large intestinal tissue thereby affecting the composition of the mouse intestinal microflora.
Fluorescent in situ hybridisation analysis
The 250 bp PCR product from the butyrate kinase gene was labelled with digoxigenin-11-2′-deoxy-uridine-5′-triphosphate (DIG-11-UTP) under the PCR conditions described earlier but with the addition of 0·5 μl (1 nmol/μl) DIG-11-UTP (Roche Diagnostics GmbH, Mannheim, Germany).
The slides containing large-intestinal sections were incubated at 56°C overnight. Sections were then deparaffinised in xylene for 10 min while shaking at 60°C. This process was repeated three times and then the slides were washed twice with 100 % ethanol for 5 min each time. The slides were then incubated in 2 × saline sodium citrate (SSC) buffer at room temperature for 2 min. This process was repeated three times and then the slides were dehydrated for 1 min in 70 %, then 85 % and finally 100 % (v/v) cold ethanol (aqueous solution) at − 20°C. The slides were then incubated for 1 h in tri(hydroxymethyl)-aminomethane (100 mmol/l), EDTA (50 mmol/l) buffer containing lysozyme (0·1 mg/ml), at 37°C, to dissolve the bacterial wall. Then the slides were dehydrated again in cold graded cold ethanol solutions as before.
The slides were warmed in a slide plate for 5 min at 37°C following 5 min incubation, at 75°C, in 60 ml denaturation buffer containing 49 ml 70 % (v/v) formamide, 7 ml 20 × SSC and 14 ml sterile water. The slides were washed in graded cold ethanol solutions as before.
Hybridisation buffer was prepared to contain 250 μl 70 % formamide, 100 μl 10 × SSC phosphate buffer, 100 μl 50 % (w/v) dextran sulfate and 20 μl of salmon sperm DNA (10 mg/ml). The hybridisation buffer was warmed at 70°C for 10 min and 9 μl warm hybridisation buffer and 1 μl of probe (25 ng/μl) were applied to the treated slides and then covered with cover slips and sealed with Fixogum rubber cement. The slides were then incubated at 37°C in an incubator overnight.
The rubber cement was removed and the slides were washed in 20 × SSC at 72°C and twice for 1 min in PBS containing Tween 20 (50 μl Tween 20 in 200 ml PBS). The slides were incubated for 10 min at room temperature with Reagent A from a Spot-light CISH translocation detection kit (Zymed Laboratories Inc., San Francisco, CA, USA). Anti-digoxigenin-rhodamine (50 μl; from a stock solution containing anti-digoxigenin-rhodamine (200 μl/ml) with 10 % (v/v) 1:250 horse serum) was added to the slides and incubated for 40 min at 37°C. The slides were then washed twice in PBS–Tween 20 for 2 min each time and the slides were dried. Finally, slides were counterstained with 4′,6-diamidino-2-phenylindole (1 mg/ml) in anti-fade solution and examined with an Olympus U-RFL-T microscope (Olympus Europe GmbH, Hamburg, Germany) suitable for FISH analyses.
Statistical analysis
Every experiment was conducted under a definite experimental design and specific statistical models were used to discern differences among treatments. Values are expressed as mean values with their standard errors. Data analyses were performed by t test or by the Tukey–Kramer test. Differences were considered significant at P < 0·05.
Results
Butyrate detection and quantification
A diluted fresh faecal sample was grown in IMM broth prepared at a wide range of different pH (5·0, 5·5, 6·0, 6·5, 7·0, 7·5, 8·0, 9·6) and incubated for 3 weeks. Butyrate concentration was determined weekly by GC. The control consisted of IMM solution without faeces prepared at pH 9·6. Different pH were used to mimic the intraluminal pH of the different compartments of the gastrointestinal tract. The highly acid pH in the stomach is changed to about pH 6 in the duodenum and increases to about pH 7·4 in the terminal ileum and then the pH drops to 5·7 in the caecum, but again gradually increases, reaching pH 6·7 in the rectum. Therefore we concentrated on pH from 5·0 to 9·6. The highest amount of butyrate was recorded in samples incubated at pH 5·0, 5·5 and 6·0 following 2 weeks of incubation, relative to the control; however, the optimal pH for butyrate production was 6·0 (Table 1). We elected the optimal and not the maximal butyrate secretion, i.e. at pH 6·0 a steady butyrate secretion is achieved already at 1, 2, 3 and 4 h. The faecal culture producing the optimal amount of butyrate was spread on an IMM agar plate and colonies were isolated.
Table 1 Determination of optimal intestinal microflora medium (IMM) conditions for faecal butyrate production† (Mean values with their standard errors)
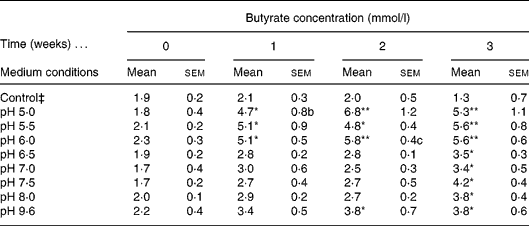
Mean value was significantly different from that of the control condition: * P < 0·05, ** P < 0·01 (Tukey–Kramer test).
† Four different independent experiments were conducted.
‡ Control culture contained IMM at pH 9 without faecal bacterial samples.
Bacterial characterisation of high-butyrate producing selected isolate
Gram staining of the bacterial isolates demonstrated that it is a Gram-positive coccus. The PCR product obtained following amplification with the 16S rRNA sequence was 1000 bp in size. The 1000 bp sequence exhibited phylogenetic identity to E. durans category (GenBank accession no. DSM20; 99 % probability).
Since the selected bacterium produced butyrate, it was tested for the expression of genes encoding one of the key enzymes participating in the butyrate biosynthesis pathway, butyrate kinase. Using the primers described earlier (p. 726), a fragment of 250 bp was amplified and sequenced (data not shown). The sequence was found to have100 % similarity (at the amplified sequence) to the gene encoding the bacterial enzyme butyrate kinase expressed in the genome of other additional butyrogenic bacteria such as Clostridium (Duncan et al. Reference Duncan, Barcenilla, Stewart, Pryde and Flint2002). The selected bacterial isolate was devoid of gelatinase activity (Franz et al. Reference Franz, Holzapfel and Stiles1999), as obtained by gelatin-zymography assay (data not shown).
As indicated in Fig. 1 (A), the selected bacterial isolate was able to grow at pH 2 and 3 for 4 h. Although the number of surviving bacteria following 1 h of incubation at pH 2 and 3 was significantly lower than in the control at pH 6, a significant number still survived and kept growing after 4 h incubation under the acidic conditions. It fulfilled additional probiotic criteria such as resistance to bile acids (Fig. 1 (B)), and survived 4 h in 0·5 % ox-gall (bile acid mixture).

Fig. 1 (A) The effect of pH on bacterial growth rate. (–♦–), Control (pH 6); (–□–), pH 2; (–Δ–), pH 3. (B) The effect of bile acids on bacterial growth rate. (–♦–), Control (–□–), bile acids. Data are means of four independent cultures, with standard errors represented by vertical bars. Mean values were significantly different: * P < 0·05, ** P < 0·001.
Percentage adhesion of E. durans to Caco-2 cells was compared with the well-known probiotic strains L. rhamnosus GG and L. casei Shirota, which are considered to be high- and low-adhesive strains, respectively (Tuomola & Salminen, Reference Tuomola and Salminen1998). The percentage of Caco-2 cells in each field with adhering bacteria and demonstrated that the number of adhered E. durans cells was comparable with the highly adhesive strain L. rhamnosus GG but significantly higher (P < 0·005 as calculated by the Tukey–Kramer test) than the low-adhesive strain L. casei Shirota (data not shown).
The selected bacterial isolate was devoid of gelatinase activity according to the gelatin-zymography assay (data not shown).
Effect of Enterococcus durans on dextran sodium sulfate-induced colitis
Acute colitis was induced in mice via administration of DSS in the drinking water. At the end of the experiment, significant differences were observed among the different groups in terms of body-weight gain and extent of rectal bleeding, measurements that were all scored as DAI. Daily administration of 1 × 108 cfu E. durans/g chow to DSS-treated mice induced a significant lowering effect in DAI, i.e. E. durans prevented weight loss in the DSS-treated mice and concomitantly the administered bacteria prevented colonic bleeding. In contrast, the food-grade bacteria L. delbrueckii subsp. lactis ATCC 4797 was completely ineffective at ameliorating DAI in mice treated with DSS.
Histological analyses demonstrated that DSS-induced colitis caused severe mucosal damage that included massive necrosis of the crypt cells (see Fig. 2 (B)); the control mice showed well-organised colonic mucosa (Fig. 2 (A)). A significant beneficial effect was observed following dietary administration of E. durans (see Fig. 2 (D)); it significantly inhibited the histological damage induced by DSS; however, this was not the case for L. delbrueckii subsp. lactis ATCC 4797 (see Fig. 2 (C)). The DAI (Fig. 3 (A)) mirrored the histological conditions of the different mouse groups. Histological scores for signs of colitis are summarised in Fig. 3 (C) and mirror the photomicrographs presented in Fig. 2. As shown in Fig. 3 (B), the intestinal inflammation induced by DSS and measured in terms of MPO activity was significantly ameliorated by oral administration of the selected high-butyrate-secreting bacteria E. durans. In contrast, L. delbrueckii subsp. lactis ATCC 4797 was completely ineffective in this regard and thus was further used as control bacteria.

Fig. 2 Intestinal histology of murine sections of the distal colon shown by haematoxylin and eosin staining. (A) Control mice; (B) dextran sodium sulfate (DSS)-treated mice; (C) DSS-treated mice fed 1 × 108 colony-forming units (cfu) Lactobacillus delbrueckii subsp. lactis/g feed; (D) DSS-treated mice fed 1 × 108 cfu Enterococcus durans/g feed.

Fig. 3 Effect of Enterococcus durans (ED) on dextran sodium sulfate (DSS)-induced colitis. (A) Effect of ED on DSS-induced colitis, as reflected by disease activity index (DAI). Scores were defined as: loss in body weight, 0 = no loss; 1 = 5 to 10 % loss; 2 = 11 to 15 % loss; 3 = 16 to 20 % loss; 4 = over 20 % loss; haemoccult, 0 = no blood; 2 = positive; 3–4 = gross blood. (B) Effect of ED on DSS-induced colitis, as reflected by myeloperoxidase (MPO) activity. (C) Effect of ED on DSS-induced colitis, as reflected by histological grade. Histological findings were also graded according to stage of intestinal inflammation. The grading scale is described on p. 730.a,b,c Mean values with unlike letters were significantly different as calculated by the Tukey–Kramer test (P < 0·01).
To verify the anti-inflammatory role of E. durans in DSS-induced colitis, we measured colonic cDNA expression of the inflammatory cytokines IL-1β and TNF-α in control, DSS-treated animals or DSS-treated animals fed with E. durans or L. delbrueckii as control bacteria (Fig. 4). In DSS-treated animals, high IL-1β and TNF-α cDNA levels were observed. In E. durans-fed animals a striking down regulation in cytokine transcription was observed. In contrast, in L. delbrueckii-fed animals, the concentrations of IL-1β and TNF-α were similar to those recorded in DSS-treated animals.

Fig. 4 Effect of Enterococcus durans (ED) on dextran sodium sulfate (DSS)-induced colitis on colonic IL-1β and TNF-α expression. RNA extracts were obtained from colonic sections of control mice, DSS-treated mice, DSS-treated mice fed 1 × 108 colony-forming units (cfu) Lactobacillus delbrueckii (LD)/g feed or DSS-treated mice fed 1 × 108 cfu ED/g feed. Colonic RNA extracts were analysed for IL-1β and TNF-α cDNA expression by RT-PCR and compared with glyceraldehyde-3-phosphate dehydrogenase (G3PDH) transcription (a constitutive enzyme of mammalian cells, a catalytic enzyme involved in glycolysis, and therefore used as a loading control). One representative experiment from three identical ones is shown.
Localisation of butyrate-producing bacteria in colon by fluorescent in situ hybridisation method
FISH analysis was used to detect the presence of E. durans in different colonic and caecal tissue sections of mice from different experimental groups. Probes specific for the bacterial butyrate kinase gene were used for PCR amplification as part of the characterisation process of E. durans. Fig. 5 (D) exemplifies a colonic section of DSS-treated mice fed with E. durans. These mice revealed a very high concentration of butyrate kinase-positive enterobacterial colonies relative to control mice (Fig. 5 (A)) or to mice treated with DSS (Fig. 5 (B)) or to those treated with DSS and fed L. delbrueckii subsp. lactis (Fig. 5 (C)). The results indicate that E. durans species do not normally populate the large intestine of mice; however, when they are orally administered they can colonise the large intestine, proliferate and inhibit DSS-induced colitis.

Fig. 5 Localisation of Enterococcus durans butyrate-producing bacteria by fluorescent in situ hybridisation (FISH) analyses for the bacterial butyrate kinase gene in colonic tissue areas of mice. (A) Control mice; (B) dextran sodium sulfate (DSS)-treated mice; (C) DSS-treated mice fed 1 × 108 colony-forming units (cfu) Lactobacillus delbrueckii subsp. lactis/g feed; (D) DSS-treated mice fed 1 × 108 cfu E. durans/g feed. FISH analyses utilising the butyrate kinase gene probe allowed us the identification of E. durans tightly attached to the mouse colonic mucosa, shown as bright spotted fluorescent staining (D). The blue staining represents the colonocyte's nuclei. Only in colonic mucosa from mice fed E. durans was the presence of the bacteria evident (D). In all other sections, from control mice (A), DSS-treated mice (B), DSS-treated mice fed with 1 × 108 cfu L. delbrueckii/g feed (C), no E. durans was identified.
Discussion
Butyrate arises from microbial fermentation, it is important for the energy metabolism and normal development of colonic epithelial cells and has a mainly protective role in colonic disease (Avivi-Green et al. Reference Avivi-Green, Madar and Schwartz2000, Reference Avivi-Green, Polak-Charcon, Madar and Schwartz2002). Butyrate has anti-inflammatory effects that result from inhibition of activation of the transcription factor NFκB, and consequent reduced formation of pro-inflammatory cytokines (Luhrs et al. Reference Luhrs, Gerke, Boxberger, Backhaus, Melcher, Scheppach and Menzel2001, Reference Luhrs, Gerke, Muller, Melcher, Schauber, Boxberge, Scheppach and Menzel2002). These interactions have important consequences for the health of the colonic epithelium. Recent studies have concentrated on combining molecular and cultural approaches to investigate the dominant butyrate-producing bacteria of the human colon and have identified a large number of butyrate-producing isolates based on sequencing and restriction fragment length polymorphism analyses of 16S rRNA genes (Barcenilla et al. Reference Barcenilla, Pryde, Martin, Duncan, Stewart, Henderson and Flint2000).
In the present study, using a cultural-restriction technique, a novel butyrate-producing bacterium that inhabits the human large intestine was identified. We characterised one bacterial isolate from a human faecal sample harvested from a vegetarian volunteer and showed that this bacterium can synthesise significant amounts of butyrate under anaerobic conditions at pH 6·0. We chose pH 6·0 and not pH 5·0, the pH that allowed peak butyrate production, since pH 6·0 is the ambient pH found in the caecum and proximal large intestine, and at pH 6·0 additional bacteria which are part of the large-intestinal flora are functionally active (Collado et al. Reference Collado, Hernandez and Sanz2005). The level of production of butyrate at the chosen pH seems to be similar to the activity reported for butyrogenic bacteria (Duncan et al. Reference Duncan, Barcenilla, Stewart, Pryde and Flint2002; Walker et al. Reference Walker, Duncan, McWilliam Leitch, Child and Flint2005).
The purpose of the present research was to find a way to create microanaerobic conditions and be able to isolate butyrate-producing bacteria from human faeces. These non-restrictive anaerobic conditions should allow their putative use in the food industry. To exert a beneficial effect in the gut, probiotic cultures must survive passage through the stomach and be tolerant to the concentrations of bile present in the small intestine (Otles et al. Reference Otles, Cagindi and Akcicek2003) in order to reach the colon. Survival at pH 3·0 for 2 h and growth in medium containing 500–1000 mg bile acids per litre are considered standard acid and bile tolerance in probiotic cultures (Snelling, Reference Snelling2005). The present bacterial isolate fulfilled these requirements. Both of these traits are prerequisites for efficacious probiotics, and variability in survival rates may help explain the controversy over their beneficial aspects.
Primary characterisation analyses demonstrated that the isolated bacteria are Gram-positive cocci. We tested whether the bacterial isolate could produce antimicrobial materials such as bacteriocins in analogy to other probiotic bacteria (Edelman et al. Reference Edelman, Bauer, Khanwani, Tait, Trepel, Karp, Nemieboka, Chung and Van Echo2003; Collado et al. Reference Collado, Hernandez and Sanz2005) and found that they do not (data not shown). There are additional pathways via which putative probiotic bacteria can act against pathogens. A case in point is the stimulation of cytokine secretion (Erickson & Hubbard, Reference Erickson and Hubbard2000; Kalliomaki & Isolauri, Reference Kalliomaki and Isolauri2004; Schultz et al. Reference Schultz, Strauch, Linde, Watzl, Obermeier, Gottl, Dunger, Grunwald, Scholmerich and Rath2004) or the production of butyrate which is used as a major energy source for the colon cells (Frankel et al. Reference Frankel, Zhang, Singh, Klurfeld, Don, Sakata, Modlin and Rombeau1994; Duncan et al. Reference Duncan, Barcenilla, Stewart, Pryde and Flint2002) and as an inhibitor of pro-inflammatory factors (D'Argenio & Mazzacca, Reference D'Argenio and Mazzacca1999; Luhrs et al. Reference Luhrs, Gerke, Boxberger, Backhaus, Melcher, Scheppach and Menzel2001, Reference Luhrs, Gerke, Muller, Melcher, Schauber, Boxberge, Scheppach and Menzel2002). In the present study, the bacterial isolate demonstrated adherence to Caco-2 cells in vitro. In the colon, the bacteria need to adhere to the epithelial cells and form active colonies. The colonised bacteria can influence the host via their metabolic activity and can compete with pathogenic bacteria (Jacobsen et al. Reference Jacobsen, Rosenfeldt Nielsen, Hayford, Moller, Michaelsen, Paerregaard, Sandstrom, Tvede and Jakobsen1999).
We genetically characterised the bacterial isolate by PCR amplification of the 16S rRNA. The sequence obtained following 16S rRNA amplification was 99 % identical to an E. durans isolate in the GeneBank. Enterococcus bacteria such as E. durans, E. faecalis, E. faecium, E. hirae, E. casseliflavus and E. gallinarum are known to be significant for the gastrointestinal ecology as well as for that of some foods (Klein, Reference Klein2003). Enterococcus bacteria exist in soil, water and in human and animal intestines (105–107 cfu/g faeces). E. faecalis and E. faecium are the most dominant strains found in the human intestine. Enterococcus strains are already known as probiotic bacteria and, to a certain extent, are used to treat gastroenteritis in human patients and animals (Franz et al. Reference Franz, Holzapfel and Stiles1999). In this regard, Pereira & Gibson (Reference Pereira and Gibson2002) have recently identified E. durans DSM 20 633 as a potential probiotic isolated from faecal material.
Sequencing the 250 bp PCR product obtained following amplification with specific primers to bacterial butyrate kinase indicated that in the isolated butyrate-producing E. durans this gene exists in its genome, as judged from 100 % identity of the 250 bp amplified to the respective sequence in the gene present in the anaerobic butyrate-producing strain C. acetobutylicum. Two key enzymes have been found to be involved in the last stage of the butyrate-synthesising process in bacteria; butyrate kinase and butyryl CoA:acetate CoA transferase (Duncan et al. Reference Duncan, Barcenilla, Stewart, Pryde and Flint2002, Reference Duncan, Scott, Ramsay, Harmsen, Welling, Stewart and Flint2003, Reference Duncan, Holtrop, Lobley, Calder, Stewart and Flint2004a , Reference Duncan, Louis and Flint b ; Louis et al. Reference Louis, Duncan, McCrae, Millar, Jackson and Flint2004). The butyryl CoA:acetate-CoA transferase gene has been found in most butyrate-producing bacteria from the human intestine, whereas butyrate kinase is less common (Duncan et al. Reference Duncan, Barcenilla, Stewart, Pryde and Flint2002, Reference Duncan, Holtrop, Lobley, Calder, Stewart and Flint2004a , Reference Duncan, Louis and Flint b ; Louis et al. Reference Louis, Duncan, McCrae, Millar, Jackson and Flint2004). Nonetheless, both pathways can be expressed at once. The significance of these metabolic differences has yet to be established.
An additional important issue to be addressed relates to bacterial safety for human use. We analysed whether the isolated E. durans has a plasmid and found none. This suggests that if the bacteria become resistant to any antibiotics, transfer of this trait to other bacteria would be impossible. We found that the bacterial isolate cannot survive in the presence of ampicillin, tetracycline and chloramphenicol. We also examined the ability of the bacterial isolate to synthesise gelatinase, an activity associated with harmful factors such as adhesins, invasins and haemolysin (Franz et al. Reference Franz, Holzapfel and Stiles1999; Kawalec et al. Reference Kawalec, Potempa, Moon, Travis and Murray2005). We showed that E. durans isolated in the present study does not bear gelatinase activity. Cumulatively these qualities further support the view that the bacterial isolate can be safe for human use.
An additional important finding of the present study relates to the ability of the bacterial isolate to prevent DSS-induced colitis. We tested the effect of E. durans on a murine model of ulcerative colitis simultaneously induced by DSS. Several animal models of experimental ulcerative colitis have been described and, of these, colitis induced in mice by oral administration of DSS has been widely used because of its many similarities to human ulcerative colitis (Cooper et al. Reference Cooper, Murthy, Shah and Sedergran1993). E. durans was able to prevent disease induction almost completely as reflected by DAI, MPO activity and histological grade, while L. delbrueckii subsp. lactis ATCC 4797 bacteria was completely ineffective in this regard.
Using a preventive setup in the present study, we demonstrated that following daily feeding of DSS-treated mice with 1 × 108 E. durans per g feed, there is significant inhibition in the development of chronic experimental colitis. The mechanism by which E. durans exerts its protection in DSS-induced colitis may reside in the bacteria's effect on immune-system regulation, or on the ability of the E. durans isolate to produce high concentrations of butyrate. Butyrate's role in preventing and/or ameliorating conditions such as ulcerative colitis has been studied (Hallert et al. Reference Hallert, Bjorck, Nyman, Pousette, Granno and Svensson2003; Vernia et al. Reference Vernia, Annese and Bresci2003; Galvez et al. Reference Galvez, Rodriguez-Cabezas and Zarzuelo2005). It has been proposed that an inadequate supply of energy to colonocytes, 70 % of which is normally obtained from butyrate, can be a causative factor in colitis. DSS, which inhibits butyrate oxidation without affecting glucose metabolism, induces colitis when given orally to mice (Ahmad et al. Reference Ahmad, Krishnan, Ramakrishna, Mathan, Pulimood and Murthy2000), and it has been proposed that sulfide toxicity results largely from inhibition of the butyrate energy supply to colonocytes. It is surmised that DSS changes in epithelial cell metabolism are secondary to mucosal inflammation. E. durans can supply sufficient butyrate concentrations to counteract the effect of DSS or inhibit intestinal mucosal inflammation, or both concomitantly. Present work in our laboratory is addressing each pathway and its significance. We conclude that E. durans prevents the induction of ulcerative colitis by DSS, significantly improving the clinical conditions. This could have important biological consequences for human health.
We used FISH analysis in order to localise the fate of E. durans in the mouse intestinal tissue. We used a 250 bp probe obtained by PCR amplification of the bacterial butyrate kinase gene to localise the E. durans in the mice colonic tissue. A high concentration of fluorescent dots on the colonic tissue surfaces of mice fed E. durans were obtained, whereas no such fluorescent dots were detected in control mice or in those fed with L. delbrueckii. This suggests that E. durans does not normally populate the mouse caecal and large-intestinal tissue surface; however, when these bacteria colonise the rodent intestine they are not only harmless to the mice, but also prevent DSS-induced colitis.
In conclusion, we described in the present paper the characterisation of a bacterial isolate exhibiting 99 % homology to E. durans and expressing typical probiotic features. Oral administration of the bacterial isolate prevented and/or ameliorated experimental colitis, suggesting that this isolate provides a unique oral anti-inflammatory tool that deserves further studies specifically addressing the pathogenesis and treatment of human inflammatory bowel disease.Additional in vivo and in vitro studies are needed, to determine the mechanism by which this bacterial isolate exerts its preventive effect on ulcerative colitis. Moreover, its influence on the indigenous microflora and overall metabolic activity of the gut should be further assessed in vitro in mixed-culture and mixed-substrate environments before the design of any clinical intervention trials. We believe that the putative probiotic bacteria described in the present paper could potentially make a significant contribution to the food and medicinal industries.
Acknowledgements
This study was supported in part by Danone Research Center, Israel.






