



Introduction
Protein misfolding and aggregation are associated with a wide variety of human diseases (Hardy and Selkoe, Reference Hardy and Selkoe2002; Eisenberg and Jucker, Reference Eisenberg and Jucker2012; Jucker and Walker, Reference Jucker and Walker2013; Knowles, Vendruscolo and Dobson, Reference Knowles, Vendruscolo and Dobson2014; Chiti and Dobson, Reference Chiti and Dobson2017). These diseases have been linked to a progressive failure of the protein homoeostasis system, which controls protein folding, trafficking and degradation (Balch et al., Reference Balch, Morimoto, Dillin and Kelly2008; Kim et al., Reference Kim, Hipp, Bracher, Hayer-Hartl and Ulrich Hartl2013). The capacity of this system has been shown to decline with age both in model organisms and in humans, which can result in the accumulation of aberrant protein aggregates (Labbadia and Morimoto, Reference Labbadia and Morimoto2015). Neurons appear to be particularly vulnerable to such events (Freer et al., Reference Freer, Sormanni, Vecchi, Ciryam, Dobson and Vendruscolo2016; Surmeier et al., Reference Surmeier, Obeso and Halliday2017; Luna et al., Reference Luna, Decker, Riddle, Caputo, Zhang, Cole, Caswell, Xie, Lee and Luk2018; Fu et al., Reference Fu, Possenti, Freer, Nakano, Hernandez Villegas, Tang, Cauhy, Lassus, Chen, Fowler, Figueroa, Huey, Johnson, Vendruscolo and Duff2019), and the vast majority of neurodegenerative diseases are accompanied by the formation of protein deposits, including those of amyloid-β (Aβ) and tau in Alzheimer's disease (AD), and of α-synuclein in Parkinson's disease (PD) (Hardy and Selkoe, Reference Hardy and Selkoe2002; Eisenberg and Jucker, Reference Eisenberg and Jucker2012; Jucker and Walker, Reference Jucker and Walker2013; Knowles et al., Reference Knowles, Vendruscolo and Dobson2014; Chiti and Dobson, Reference Chiti and Dobson2017).
Although mature Aβ fibrils are in many cases relatively inert, studies in cell culture and animal models have revealed that misfolded oligomers, which are formed during the process of fibril formation, possess cytotoxic properties (Bucciantini et al., Reference Bucciantini, Giannoni, Chiti, Baroni, Formigli, Zurdo, Taddei, Ramponi, Dobson and Stefani2002; Baglioni et al., Reference Baglioni, Casamenti, Bucciantini, Luheshi, Taddei, Chiti, Dobson and Stefani2006; Haass and Selkoe, Reference Haass and Selkoe2007; Bemporad and Chiti, Reference Bemporad and Chiti2012; Benilova et al., Reference Benilova, Karran and De Strooper2012; Chiti and Dobson, Reference Chiti and Dobson2017). Several mechanisms have been proposed to account for the toxicity of these oligomers, including disruption of lipid membranes, transcriptional deregulation, mitochondrial dysfunction, oxidative stress, proteasome inhibition and disruption of synaptic plasticity (Haass and Selkoe, Reference Haass and Selkoe2007; Benilova et al., Reference Benilova, Karran and De Strooper2012; Roberts and Brown, Reference Roberts and Brown2015; De et al., Reference De, Wirthensohn, Flagmeier, Hughes, Aprile, Ruggeri, Whiten, Emin, Xia, Varela, Sormanni, Kundel, Knowles, Dobson, Bryant, Vendruscolo and Klenerman2019). Some of these mechanisms affect components of the protein homoeostasis system, resulting in a collapse of protein quality control and a further increase in protein misfolding and toxicity (Labbadia and Morimoto, Reference Labbadia and Morimoto2015). On the whole, however, the molecular mechanisms whereby protein misfolding gives rise to toxicity are not yet understood to the extent needed to enable the development of disease-modifying treatments for the vast majority of these disorders.
In this context, in vivo models can both increase our understanding of the mechanisms of disease and provide a platform for drug screening and preclinical development (Dawson et al., Reference Dawson, Golde and Lagier-Tourenne2018; Götz et al., Reference Götz, Bodea and Goedert2018). A variety of model organisms has been exploited to probe protein misfolding diseases, ranging from simple unicellular systems such as yeast, to more complex ones such as rodents and primates. Simple organisms lack specific aspects of human biology, but can be faster and more cost-effective to work with, making them particularly suitable for high-throughput screens, and for the study of highly conserved cellular pathways (Khurana and Lindquist, Reference Khurana and Lindquist2010; Rincon-Limas et al., Reference Rincon-Limas, Jensen and Fernandez-Funez2012; Sin et al., Reference Sin, Michels and Nollen2014). More complex organisms can recapitulate human disease phenotypes better, and are typically used in preclinical studies to examine efficacy and safety prior to testing in humans (Jucker, Reference Jucker2010; Sasaguri et al., Reference Sasaguri, Nilsson, Hashimoto, Nagata, Saito, De Strooper, Hardy, Vassar, Winblad and Saido2017).
In general, in vivo models of disease can be studied at different levels. Organismal and cellular phenotypes can be related to the aggregation states of disease-associated proteins at the molecular level by biochemical and biophysical tools (Table 1). Correspondingly, at the system level, disrupted molecular pathways can be comprehensively monitored by genomics, transcriptomics and proteomics techniques (Barabási et al., Reference Barabási, Gulbahce and Loscalzo2011). With these approaches, the association of a perturbation (e.g. the expression of a mutant gene) with a phenotype (i.e. an aspect of the disease) is translated into knowledge of specific biochemical processes associated with the disease under investigation. Finally, a comparison with in vitro studies can contribute to deciphering the molecular mechanisms of these processes, and represents a powerful strategy for revealing the molecular origins of a disease, and for identifying possible targets for therapeutic interventions (Knowles et al., Reference Knowles, Vendruscolo and Dobson2014; Chiti and Dobson, Reference Chiti and Dobson2017).
Table 1. Commonly used biophysical methods for the study of protein misfolding in vivo and in vitro
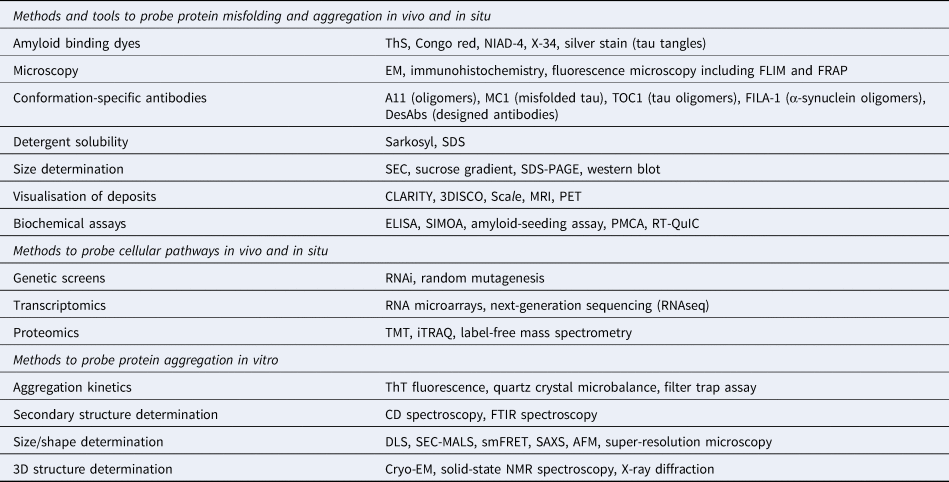
The application of these methods to study protein aggregation and the associated references are mentioned throughout the main text.
Here, we provide an overview of the analysis of protein misfolding in transgenic models of AD and PD based on the transgenic expression of Aβ or tau, and of α-synuclein, respectively. We furthermore discuss methodological challenges in developing biophysical tools to probe in vivo the aggregation process and the resulting aggregate species, which are required to provide quantitative assessments of cause–effect relationships in disease and of the efficacy of potential therapies.
Determination of protein aggregation mechanisms using in vitro biophysical methods
Biophysical studies of protein aggregation in vitro enable the identification of possible molecular mechanisms of protein aggregation in vivo and the corresponding neurodegenerative processes (Knowles et al., Reference Knowles, Vendruscolo and Dobson2014; Chiti and Dobson, Reference Chiti and Dobson2017). This approach is powerful not only because in vitro systems are amenable to a wider range of technologies compared to in vivo systems (Table 1) but also because experimental conditions can be carefully controlled, and the effects of specific perturbations can be examined, e.g. with the introduction of cellular components whose relevance has been revealed by in vivo studies.
In particular, the kinetics of the protein aggregation process can be measured quantitatively in vitro, leading to a detailed mechanistic understanding of the process itself. Protein aggregation over time is typically monitored by measuring the fluorescence of the amyloid-sensitive dye thioflavin T (ThT). The analysis of the concentration dependence of the time courses of the ThT signal leads to a description of the microscopic steps in the aggregation process and the associated rate constants (Fig. 1a) (Knowles et al., Reference Knowles, Waudby, Devlin, Cohen, Aguzzi, Vendruscolo, Terentjev, Welland and Dobson2009; Cohen et al., Reference Cohen, Vendruscolo, Dobson and Knowles2012; Meisl et al., Reference Meisl, Kirkegaard, Arosio, Michaels, Vendruscolo, Dobson, Linse and Knowles2016a; Michaels et al., Reference Michaels, Liu, Meisl and Knowles2017). Aggregation reactions that start from purely monomeric species require primary nucleation events, which involve the assembly of some monomers into disordered oligomers, which can then undergo a structural conversion into forms that have the capacity to grow by elongation through the addition of more monomers (Michaels et al., Reference Michaels, Šarić, Curk, Bernfur, Arosio, Meisl, Dear, Cohen, Dobson, Vendruscolo, Linse and Knowles2020). When fibrils are formed, secondary processes play a significant role in a variety of systems, and in some cases can dominate the overall aggregation behaviour (Cohen et al., Reference Cohen, Linse, Luheshi, Hellstrand, White, Rajah, Otzen, Vendruscolo, Dobson and Knowles2013b; Meisl et al., Reference Meisl, Yang, Hellstrand, Frohm, Kirkegaard, Cohen, Dobson, Linse and Knowles2014). These processes are dependent on the concentration of the existing amyloid aggregates, and include fibril fragmentation as well as secondary nucleation of monomers on fibril surfaces (Fig. 1b). Secondary processes represent a feed-forward loop and cause an exponential proliferation of fibrils (Cohen et al., Reference Cohen, Linse, Luheshi, Hellstrand, White, Rajah, Otzen, Vendruscolo, Dobson and Knowles2013b; Meisl et al., Reference Meisl, Yang, Hellstrand, Frohm, Kirkegaard, Cohen, Dobson, Linse and Knowles2014). It is of great importance to understand the relative contributions of the microscopic steps underlying the aggregation process in the context of disease, especially given that potentially toxic oligomers may arise from both primary and secondary nucleation.

Fig. 1. Mechanisms of amyloid formation in vitro. (a) In vitro ThT aggregation assay of Aβ42 at different starting monomer concentrations. The curves are fitted using a model that includes both primary and secondary nucleation. Figure adapted from Cohen et al. (Reference Cohen, Linse, Luheshi, Hellstrand, White, Rajah, Otzen, Vendruscolo, Dobson and Knowles2013b). (b) Schematic representation of the kinetic model of fibril formation including primary and secondary processes. Soluble monomeric forms of proteins undergo primary nucleation to generate oligomeric species that have the potential to convert into fibrils. The formation of fibrils is significantly enhanced by secondary processes that enable aggregates to proliferate by the further association of soluble protein molecules. The latter processes include fragmentation, which generates new fibril ends at which growth occurs, and surface catalysed nucleation, in which fibril surface functions as a template for the generation of new oligomeric and fibrillar species. Figure based on Cohen et al. (Reference Cohen, Linse, Luheshi, Hellstrand, White, Rajah, Otzen, Vendruscolo, Dobson and Knowles2013b). (c) ThT aggregation assay of α-synuclein in the presence of different concentrations of dimyristoylphosphatidylserine (DMPS) vesicles (black, 60 μM; purple, 120 μM; dark blue, 180 μM; light blue, 240 μM; dark green, 300 μM; light green, 450 μM; yellow, 600 μM; orange, 1200 μM). Figure adapted from Galvagnion et al. (Reference Galvagnion, Buell, Meisl, Michaels, Vendruscolo, Knowles and Dobson2015). (d) Schematic representation of the dominating mechanism of amyloid formation by α-synuclein in the presence of small unilamellar lipid vesicles, which present an interface for nucleation to occur. Figure based on Galvagnion et al. (Reference Galvagnion, Buell, Meisl, Michaels, Vendruscolo, Knowles and Dobson2015).
We present here recent insights obtained from in vitro experiments on the aggregation mechanisms of Aβ, tau and α-synuclein, in particular those that can be correlated directly with observations made in in vivo models.
Aβ aggregation
The aggregation of Aβ into extracellular amyloid plaques is a hallmark of both the familial (early-onset) and sporadic (late-onset) forms of AD. The involvement of amyloid precursor protein (APP) and its sequential processing into Aβ by β-secretase and γ-secretase (Fig. 2a) is well established through the amyloid hypothesis (Selkoe and Hardy, Reference Selkoe and Hardy2016). In particular, mutations in APP or in the presenilin (PSEN) subunit of γ-secretase are directly linked to familial forms of the disease, and triplication of chromosome 21, on which the APP gene is located, is associated with the development of AD in patients with Down syndrome (Karran and De Strooper, Reference Karran and De Strooper2016; Selkoe and Hardy, Reference Selkoe and Hardy2016). The relationship between Aβ aggregation and the onset and progression of AD has been debated extensively (Herrup, Reference Herrup2015; Karran and De Strooper, Reference Karran and De Strooper2016; Selkoe and Hardy, Reference Selkoe and Hardy2016), with studies suggesting that amyloid deposition may precede the onset of symptoms (Bateman et al., Reference Bateman, Xiong, Benzinger, Fagan, Goate, Fox, Marcus, Cairns, Xie, Blazey, Holtzman, Santacruz, Buckles, Oliver, Moulder, Aisen, Ghetti, Klunck, McDade, Martins, Masters, Mayeux, Ringman, Rossor, Schofield, Sperling, Salloway and Morris2012; Jack et al., Reference Jack, Knopman, Jagust, Petersen, Weiner, Aisen, Shaw, Vemuri, Wiste, Weigand, Lesnick, Pankratz, Donohue and Trojanowski2013; Villemagne et al., Reference Villemagne, Burnham, Bourgeat, Brown, Ellis, Salvado, Szoeke, Macaulay, Martins, Maruff, Ames, Rowe and Masters2013), and supporting a relationship between Aβ oligomers and cellular toxicity (Benilova et al., Reference Benilova, Karran and De Strooper2012; Chiti and Dobson, Reference Chiti and Dobson2017).
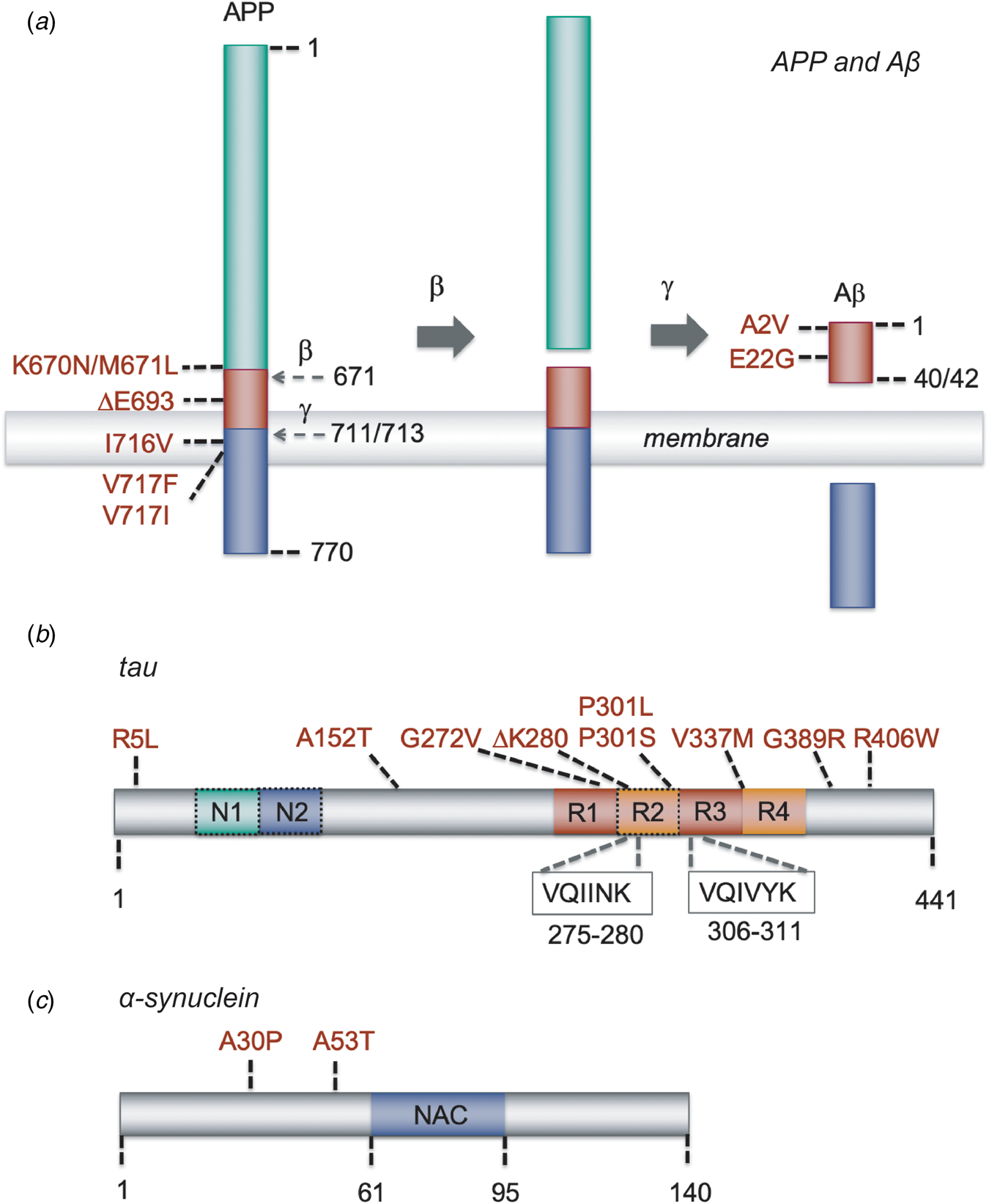
Fig. 2. Schematic representation of Aβ, tau and α-synuclein, and of the mutations that have been used in the models described in this paper. (a) APP and its sequential processing into the Aβ peptide by β-secretase and γ-secretase. Cleavage sites are indicated by the residue number at the N-terminal side of the cleavage; mutations are indicated with the residue numbers of full-length APP and those of the Aβ40/42 cleavage product. Other APP processing pathways, such as α-secretase processing, and other Aβ forms, such as Aβ39 or Aβ43, are omitted for clarity. (b) The MAPT gene encoding tau undergoes splicing to yield six possible isoforms. The full-length 2N4R tau isoform of 441 residues are shown; 1N and 0N isoforms lack the second or both N-terminal inserts, respectively, and 3R isoforms lack the second microtubule repeat (dashed boxes). The hexapeptide stretches VQIINK and VQIVYK, which have the propensity to form β-strands and are essential for the aggregation of tau, are located at the microtubule binding repeats. (c) Schematic representation of α-synuclein, containing three major regions: an N-terminal amphipathic domain with α-helical propensity where several familial mutations are located (residues 1–61), a central hydrophobic part (residues 61–95) known as the NAC region that is essential for amyloid formation, and an acidic C-terminal region (residues 95–140).
Several Aβ variants of different lengths and with different post-translational modifications exist in the human brain, with the two most common forms being of 40 (Aβ40) and 42 (Aβ42) residues (Kummer and Heneka, Reference Kummer and Heneka2014). Studying in vitro the aggregation of these different forms of Aβ, in particular with respect to oligomer formation, has led to the establishment of correlations between aggregation mechanisms and cytotoxic processes, in particular by revealing the importance of secondary nucleation in the proliferation of the aggregates (Cohen et al., Reference Cohen, Linse, Luheshi, Hellstrand, White, Rajah, Otzen, Vendruscolo, Dobson and Knowles2013b; Meisl et al., Reference Meisl, Yang, Hellstrand, Frohm, Kirkegaard, Cohen, Dobson, Linse and Knowles2014) (Fig. 1a and b). Aβ42 aggregates faster than Aβ40 in vitro, with the relative contributions of the microscopic aggregation steps being different for the two isoforms, as in particular the rate of primary nucleation is enhanced for Aβ42 (Meisl et al., Reference Meisl, Yang, Hellstrand, Frohm, Kirkegaard, Cohen, Dobson, Linse and Knowles2014). From in vitro studies, it has furthermore become clear that the two forms can affect the behaviour of each other, although the details of such interplay still remain unclear (Pauwels et al., Reference Pauwels, Williams, Morris, Jonckheere, Vandersteen, Kelly, Schymkowitz, Rousseau, Pastore, Serpell and Broersen2012; Yu-Jen and Yun-Ru, Reference Yu-Jen and Yun-Ru2014; Cukalevski et al., Reference Cukalevski, Yang, Meisl, Weininger, Bernfur, Frohm, Knowles and Linse2015; Tran et al., Reference Tran, Chang, Hsu, Wang and Guo2017).
Familial mutations linked to early-onset AD include those in the PSENs that affect the cleavage of Aβ, leading to increased levels of Aβ42 in particular. However, some mutations also occur within the Aβ sequence itself, and these have been found to cause increased aggregation propensities (see http://www.alzforum.org/mutations). A study expressing a series of Aβ42 mutants in Drosophila has found a remarkable correlation between the in vitro aggregation rates of the variants and measures of cytotoxicity, namely reduced locomotor function and lifespan, as described in the Section ‘Aβ models’ (Luheshi et al., Reference Luheshi, Tartaglia, Brorsson, Pawar, Watson, Chiti, Vendruscolo, Lomas, Dobson and Crowther2007). Using in vitro assays, further mechanistic insights have been obtained to probe which of the microscopic steps in the aggregation process are affected for the Aβ42 variants. For example, for the A2V Aβ42 mutant associated with early-onset AD, it has been shown that secondary nucleation is significantly enhanced compared to that observed for the wild-type form, potentially generating a larger flux of toxic oligomers (Meisl et al., Reference Meisl, Yang, Frohm, Knowles and Linse2016b). In addition, mutations of residues E22 and D23, which are surface-exposed, have been found to lead to enhanced secondary nucleation, whereas the elongation rates are close to that of the wild-type peptide (Yang et al., Reference Yang, Meisl, Frohm, Thulin, Knowles and Linse2018). Although the aggregation process may be affected by extrinsic factors in vivo, these findings suggest that the surface properties of Aβ fibrils play an important role in the generation of toxicity.
Given the cytotoxicity of the oligomeric forms of Aβ (Bucciantini et al., Reference Bucciantini, Giannoni, Chiti, Baroni, Formigli, Zurdo, Taddei, Ramponi, Dobson and Stefani2002; Baglioni et al., Reference Baglioni, Casamenti, Bucciantini, Luheshi, Taddei, Chiti, Dobson and Stefani2006; Haass and Selkoe, Reference Haass and Selkoe2007; Bemporad and Chiti, Reference Bemporad and Chiti2012; Benilova et al., Reference Benilova, Karran and De Strooper2012; Chiti and Dobson, Reference Chiti and Dobson2017), the structural features of these assemblies are under close investigation. Characterisation of a particular form of such oligomers by solid-state NMR spectroscopy (Table 1) has provided evidence for parallel β-sheet content (Parthasarathy et al., Reference Parthasarathy, Inoue, Xiao, Matsumura, Nabeshima, Hoshi and Ishii2015). Other structural studies on samples prepared in vitro have provided evidence for the existence of more loosely packed Aβ42 oligomers (Ahmed et al., Reference Ahmed, Davis, Aucoin, Sato, Ahuja, Aimoto, Elliott, Van Nostrand and Smith2010), as well as antiparallel β-sheet oligomers (Cerf et al., Reference Cerf, Sarroukh, Tamamizu-Kato, Breydo, Derclaye, Dufrêne, Narayanaswami, Goormaghtigh, Ruysschaert and Raussens2009; Stroud et al., Reference Stroud, Liu, Teng and Eisenberg2012; Huang et al., Reference Huang, Zimmerman, Martin, Nix, Rosenberry and Paravastu2015). A structural study of Aβ40 oligomers stabilised by zinc ions has also revealed a compresence of both parallel and antiparallel β-sheet conformations (Mannini et al., Reference Mannini, Habchi, Chia, Ruggeri, Perni, Knowles, Dobson and Vendruscolo2018). Mature Aβ42 fibrils, on the other hand, have only ever been found to adopt parallel β-sheet conformations as evidenced by recent structure determination using solid-state NMR spectroscopy (Xiao et al., Reference Xiao, Ma, McElheny, Parthasarathy, Long, Hoshi, Nussinov and Ishii2015; Colvin et al., Reference Colvin, Silvers, Ni, Can, Sergeyev, Rosay, Donovan, Michael, Wall, Linse and Griffin2016; Wälti et al., Reference Wälti, Ravotti, Arai, Glabe, Wall, Böckmann, Güntert, Meier and Riek2016) and cryo-electron microscopy (cryo-EM, Table 1) (Gremer et al., Reference Gremer, Schölzel, Schenk, Reinartz, Labahn, Ravelli, Tusche, Lopez-Iglesias, Hoyer, Heise, Willbold and Schröder2017). A conformational mapping of Aβ42 oligomers using a panel of single-domain antibodies rationally designed to scan the whole Aβ42 sequence (Aprile et al., Reference Aprile, Sormanni, Perni, Arosio, Linse, Knowles, Dobson and Vendruscolo2017) has revealed that early oligomers produced during the aggregation reaction are structurally different from those produced at later stages, and correspondingly are toxic through different mechanisms (De et al., Reference De, Wirthensohn, Flagmeier, Hughes, Aprile, Ruggeri, Whiten, Emin, Xia, Varela, Sormanni, Kundel, Knowles, Dobson, Bryant, Vendruscolo and Klenerman2019).
Lipid membranes are likely to play a central role in the aggregation process of Aβ in vivo, given that the peptide is generated from the cleavage of APP within lipid bilayers. As such, aggregation is likely to occur in the vicinity of these membranes, whether intracellularly within endosomes or lysosomes, or extracellularly near the plasma membrane. Depending on the types of lipids and the fluidity of the membranes, studies of synthetic liposomes in in vitro aggregation reactions of Aβ have led to a wide range of different results. Lipids in a gel phase have been found to have a stronger inhibitory effect on Aβ aggregation kinetics than those in liquid–crystalline bilayers (Hellstrand et al., Reference Hellstrand, Sparr and Linse2010). Furthermore, a relationship may exist between the levels of cholesterol and Aβ deposition, as the major risk factor for late-onset AD, the presence of a mutation in the ApoE gene, is involved in cholesterol metabolism (Mahley, Reference Mahley1988). Incorporation of cholesterol in lipid vesicles in aggregation assays has revealed that cholesterol does indeed promote Aβ42 aggregation (Habchi et al., Reference Habchi, Chia, Galvagnion, Michaels, Bellaiche, Ruggeri, Sanguanini, Idini, Kumita, Sparr, Linse, Dobson, Knowles and Vendruscolo2018). This effect could be due to the fact that cholesterol modifies the fluidity of the membrane, although a direct molecular interaction between cholesterol and Aβ42 has also been reported (Di Scala et al., Reference Di Scala, Chahinian, Yahi, Garmy and Fantini2014). Study of the effects of the lipid membrane composition and fluidity in more detail will be important as these properties may be altered during ageing and neurodegeneration, e.g. as a consequence of lipid oxidation, which not only modifies the lipid types but also reduces membrane fluidity (Borst et al., Reference Borst, Visser, Kouptsova and Visser2000).
Tau aggregation
Intracellular neurofibrillary tangles composed of hyperphosphorylated tau are a hallmark of AD, along with the presence of extracellular amyloid plaques. Tau deposits are observed in a family of neurodegenerative disorders collectively known as tauopathies, which in addition to AD include Pick's disease, corticobasal degeneration, progressive supranuclear palsy, argyrophilic grain disease and frontotemporal dementia (Lee et al., Reference Lee, Goedert and Trojanowski2001). In the human brain, the MAPT gene encoding tau undergoes alternative splicing leading to six possible isoforms, depending on the number of N-terminal insertions (0N, 1N or 2N) and microtubule binding repeats (3R or 4R) (Fig. 2b) (Goedert et al., Reference Goedert, Spillantini, Jakes, Rutherford and Crowther1989). Mutations in MAPT are associated with the development of several tauopathies, although specific mutations have not been reported for AD patients.
Tau was originally described as a protein that binds and stabilises microtubules (Weingarten et al., Reference Weingarten, Lockwood, Hwo and Kirschner1975). It is intrinsically disordered in its monomeric form, and has several phosphorylation sites which can be modified to regulate microtubule stability, with hyperphosphorylation being associated with disease (Wang and Mandelkow, Reference Wang and Mandelkow2016). The classical histopathological staging of the disease (Braak and Braak, Reference Braak and Braak1991), as well as recent advances in positron emission tomography (PET) imaging have revealed that tau deposition is more closely related to the symptoms of AD than is the presence of amyloid plaques (Ossenkoppele et al., Reference Ossenkoppele2016; Tosun et al., Reference Tosun, Landau, Aisen, Petersen, Mintun, Jagust and Weiner2017; Xia et al., Reference Xia, Makaretz, Caso, McGinnis, Gomperts, Sepulcre, Gomez-Isla, Hyman, Schultz, Vasdev, Johnson and Dickerson2017). However, the mechanisms by which tau causes cellular toxicity are not fully understood, and the relationship with Aβ accumulation is still under debate (Spires-Jones and Hyman, Reference Spires-Jones and Hyman2014).
The C-terminal region of tau contains the microtubule binding repeats, which include the short hexapeptide stretches VQIINK and VQIVYK that have a high propensity to form β-strands and are essential for the aggregation of tau (Wang and Mandelkow, Reference Wang and Mandelkow2016). Tau is highly hydrophilic and intrinsically disordered, although it has been shown to adopt preferentially a compact conformation in which the N- and C-terminal regions are in close proximity (Jeganathan et al., Reference Jeganathan, von Bergen, Brutlach, Steinhoff and Mandelkow2006). The N-terminal part of tau is known as the projection domain, extending away from the interaction site when tau is bound to microtubules, and away from the fibril surface in the case of fibrillar tau. Given that truncated versions of tau, or constructs comprising only the C-terminal repeat region, have increased aggregation propensity, the N-terminal region is thought to exert a protective effect (Wang and Mandelkow, Reference Wang and Mandelkow2016). Under typical in vitro conditions, however, tau does not aggregate spontaneously, but to form fibrils requires the addition of catalysts, as for example poly-anionic molecules such as heparin. The negative charges may be required to overcome the repulsion between tau molecules, each of which carries a net positive charge in the C-terminal repeat region, although heparin also induces the aggregation of phosphorylated tau with a net negative charge by an unknown mechanism (Wang and Mandelkow, Reference Wang and Mandelkow2016).
In vitro studies have been instrumental in complementing in vivo work to shed light on the role of familial mutations, splicing and phosphorylation in tau aggregation and toxicity. A key finding has been that disease-associated mutations reduce the affinity of tau for microtubules, which may increase the risk of aggregation in vivo as the same region of the protein is involved in both processes (Hong et al., Reference Hong, Zhukareva, Vogelsberg-Ragaglia, Wszolek, Reed, Miller, Geschwind, Bird, McKeel, Goate, Morris, Wilhelmsen, Schellenberg, Trojanowski and Lee1998). The six tau isoforms have been shown to vary in their aggregation propensity, with the 4R isoforms being more aggregation-prone than the 3R isoforms (Zhong et al., Reference Zhong, Congdon, Nagaraja and Kuret2012). How this finding relates to pathological conditions is unclear, however, as in AD a mixture of 3R and 4R isoforms is found in tau tangles, whereas in other tauopathies such as Pick's disease and argyrophilic grain disease either the 3R or 4R tau species dominates. A potential explanation could again be provided by post-translational modifications such as phosphorylation. Out of the 85 theoretical phosphorylation sites in the tau sequence, about 45 have been observed in tau derived from AD brains (Hanger et al., Reference Hanger, Anderton and Noble2009). In vitro assays have led to the identification of the potential kinases and phosphatases associated with various sites, and for example GSK-3 has been found to be capable of targeting many sites (Hanger et al., Reference Hanger, Anderton and Noble2009), consistent with its important role in tau toxicity as observed in a variety of animal models as outlined in the Section ‘Tau models’. In light of the complex patterns of phosphorylation, it is not surprising that conflicting results have been obtained on whether hyperphosphorylation of tau promotes or inhibits its aggregation. Hyperphosphorylated tau extracted from AD brains is able to form fibrils in vitro, whereas dephosphorylation using alkaline phosphatase inhibits aggregation into fibrils (del Alonso et al., Reference del Alonso, Zaidi, Novak, Grundke-Iqbal and Iqbal2001). Conversely, pseudo-phosphorylated constructs comprising the tau repeat region show much slower aggregation in vitro, although they may result in the formation of a larger number of oligomers (Kumar et al., Reference Kumar, Tepper, Kaniyappan, Biernat, Wegmann, Mandelkow, Müller and Mandelkow2014).
Directly and quantitatively observing the formation of oligomeric species has been challenging due to their heterogeneous and transient nature, but an elegant single-molecule Förster resonance energy transfer (smFRET, Table 1) assay has yielded important insights into the generation of tau oligomers (Shammas et al., Reference Shammas, Garcia, Kumar, Kjaergaard, Horrocks, Shivji, Eva, Knowles, Eckhard and Klenerman2015). In this assay, relatively small oligomers were detected early in the aggregation process and these were found to subsequently convert into fibrils. The familial mutants ΔK280 and P301L were not only observed to aggregate faster, but also to form a larger population of oligomers compared to the wild-type protein, the oligomers being of the same apparent size. An anti-aggregation mutant with two proline substitutions that is less toxic in mouse models (Eckermann et al., Reference Eckermann, Mocanu, Khlistunova, Biernat, Nissen, Hofmann, Schönig, Bujard, Haemisch, Mandelkow, Zhou, Rune and Mandelkow2007; Mocanu et al., Reference Mocanu, Nissen, Eckermann, Khlistunova, Biernat, Drexler, Petrova, Schonig, Bujard, Mandelkow, Zhou, Rune and Mandelkow2008) was also found to form oligomers, however. Thus, the levels of oligomeric species cannot be directly correlated with toxicity, presumably because the mutations lead to oligomers with distinct structural characteristics (Shammas et al., Reference Shammas, Garcia, Kumar, Kjaergaard, Horrocks, Shivji, Eva, Knowles, Eckhard and Klenerman2015). In a follow-up study, different types of oligomers could be distinguished by varying the buffer conditions in the smFRET system (Kjaergaard et al., Reference Kjaergaard, Dear, Kundel, Meisl, Knowles and Klenerman2018). One type was found to be sensitive to the ionic strength of the buffer, and to be in rapid exchange with tau monomers, whereas the other type was found to be more stable and presumably unable to convert to fibrils (Kjaergaard et al., Reference Kjaergaard, Dear, Kundel, Meisl, Knowles and Klenerman2018). Both species may be toxic, but it is likely that they operate via different mechanisms.
Recently, liquid–liquid phase separation has attracted attention as a biophysical phenomenon that may act as a precursor to the aggregation of several disease-related proteins (Shin and Brangwynne, Reference Shin and Brangwynne2017; Alberti and Dormann, Reference Alberti and Dormann2019). Also, the repeat region of tau has been found to condense into liquid droplets in vitro, depending on parameters such as the protein concentration, pH value and temperature (Ambadipudi et al., Reference Ambadipudi, Biernat, Riedel, Mandelkow and Zweckstetter2017). Phase separation has been observed to induce reversibly the formation of β-strands as detected by circular dichroism (CD, Table 1) spectroscopy. The absence of ThT fluorescence suggests that the phase-separated state does not consist of amyloid fibrils, however, although the addition of heparin to the droplets was found to induce amyloid-like aggregation. NMR spectroscopy (Table 1) has furthermore shown that the conformation of tau is not drastically altered in the phase-separated state, whereas paramagnetic relaxation experiments and 13C-detected experiments have provided evidence for involvement of the repeat regions and the hexapeptide motifs (Ambadipudi et al., Reference Ambadipudi, Biernat, Riedel, Mandelkow and Zweckstetter2017, Reference Ambadipudi, Reddy, Biernat, Mandelkow and Zweckstetter2019).
Full-length, phosphorylated tau has also been found to phase separate under physiological conditions and to form droplets both in vitro and in neuronal cells in culture (Wegmann et al., Reference Wegmann, Eftekharzadeh, Tepper, Zoltowska, Bennett, Dujardin, Laskowski, MacKenzie, Kamath, Commins, Vanderburg, Roe, Fan, Molliex, Hernandez-Vega, Muller, Hyman, Mandelkow, Taylor and Hyman2018). The process has been suggested to be mediated by weak hydrophobic interactions between the hexapeptide repeats, as the liquid droplets were observed to be sensitive to dissociation by urea and by 1,6-hexanediol. The N-terminal region of tau by itself was also observed to phase separate, but in this case driven by electrostatic interactions that can be disrupted by salt. The importance of electrostatics is further underscored by the effect of tau phosphorylation, which was shown to be required for efficient droplet formation. Finally, tau liquid droplets were found to convert rapidly into gels, and after prolonged incubation into non-spherical thioflavin S (ThS) positive aggregates, confirming that tau phase separation can be on pathway to aggregate formation (Wegmann et al., Reference Wegmann, Eftekharzadeh, Tepper, Zoltowska, Bennett, Dujardin, Laskowski, MacKenzie, Kamath, Commins, Vanderburg, Roe, Fan, Molliex, Hernandez-Vega, Muller, Hyman, Mandelkow, Taylor and Hyman2018). Whether phase separation drives tau aggregation in human disease remains to be determined, and it will be exciting to study this phenomenon in in vivo models, e.g. by using fluorescence lifetime imaging microscopy (FLIM, Table 1) in Caenorhabditis elegans (Laine et al., Reference Laine, Sinnige, Ma, Haack, Poudel, Gaida, Curry, Perni, Nollen, Dobson, Vendruscolo, Kaminski Schierle and Kaminski2019) (see Section ‘α-Synuclein models’).
α-Synuclein aggregation
The 140-residue protein α-synuclein (Fig. 2c) is the major component of the insoluble intracellular deposits known as Lewy bodies and Lewy neurites that are characteristically found in the brains of PD patients, as well as in those suffering from dementia with Lewy bodies (Spillantini et al., Reference Spillantini, Crowther, Jakes, Hasegawa and Goedert1998c) and multiple system atrophy (Spillantini et al., Reference Spillantini, Crowther, Jakes, Cairns, Lantos and Goedert1998a), a set of diseases collectively termed synucleinopathies. PD is associated with the specific loss of dopaminergic neurons in the substantia nigra, which results in impaired motor function, as well as a variety of other deficits including behavioural changes, sleep problems and a diminished sense of smell. Gene duplications and triplications and several point mutations in the SNCA gene, encoding α-synuclein, in hereditary forms of PD have supported a causative role for its aggregation in the disease process (Kalinderi et al., Reference Kalinderi, Bostantjopoulou and Fidani2016), although the molecular mechanisms by which this pathology occurs are still poorly understood.
Many of the biophysical properties of α-synuclein that have been established through in vitro studies have been shown to be valuable in interpreting in vivo models. In vitro aggregation assays using recombinant α-synuclein have shown that its spontaneous aggregation is negligible at neutral pH, although when preformed seeds are added the reaction can proceed rapidly (Buell et al., Reference Buell, Galvagnion, Gaspar, Sparr, Vendruscolo, Knowles, Linse and Dobson2014). At slightly acidic pH, secondary processes in which existing fibrils catalyse the formation of new ones on their surfaces start to dominate (Buell et al., Reference Buell, Galvagnion, Gaspar, Sparr, Vendruscolo, Knowles, Linse and Dobson2014). Such conditions may be relevant in cellular compartments such as lysosomes and synaptic vesicles. Furthermore, the presence of small unilamellar vesicles in the aggregation assay dramatically increases primary nucleation as α-synuclein binds to the lipid bilayers, which initiates the aggregation process (Fig. 1c and d) (Galvagnion et al., Reference Galvagnion, Buell, Meisl, Michaels, Vendruscolo, Knowles and Dobson2015, Reference Galvagnion, Brown, Ouberai, Flagmeier, Vendruscolo, Buell, Sparr and Dobson2016). In agreement with the propensity of α-synuclein to interact with lipid membranes, genetic screens have systematically identified proteins involved in vesicle trafficking to be related to α-synuclein-mediated toxicity, as described in the Section ‘Biological pathways in Parkinson's disease models’. These findings do not seem to arise simply as a result of the expression of α-synuclein in non-neuronal cell types such as yeast or C. elegans body wall muscle cells, as many of the proteins identified in the screens have been confirmed in neuronal models and in neurons from higher organisms (Cooper et al., Reference Cooper, Gitler Cashikar, Cole, Haynes, Bhullar Liu, Xu, Strathearn, Liu, Cao, Caldwell, Caldwell, Marsischky, Kolodner, Labaer, Rochet, Bonini and Lindquist2006; Gitler et al., Reference Gitler, Bevis, Shorter, Strathearn, Hamamichi, Su, Caldwell, Caldwell, Rochet, McCaffery, Barlowe and Lindquist2008; Hamamichi et al., Reference Hamamichi, Rivas, Knight, Cao, Caldwell and Caldwell2008).
Although the presence of mature Lewy body pathology does not appear to be essential for toxicity to be observed in the vast majority of in vivo models as discussed in the Section ‘α-Synuclein models’, the process of protein aggregation is likely to play a causative role in synucleinopathies, given that the more aggregation prone α-synuclein variants such as A53T (Conway et al., Reference Conway, Lee, Rochet, Ding, Williamson and Lansbury2000) or the C-terminal truncation fragment (Crowther et al., Reference Crowther, Jakes, Spillantini and MG1998) are typically associated with increased toxicity. Data on the A30P mutant are, however, more difficult to interpret, as its aggregation kinetics measured in vitro are very similar to those of the wild-type protein (Flagmeier et al., Reference Flagmeier, Meisl, Vendruscolo, Knowles, Dobson, Buell and Galvagnion2016), yet it does not readily form inclusions in yeast (Outeiro and Lindquist, Reference Outeiro and Lindquist2003) or in C. elegans body wall muscle cells (Perni et al., submitted), although A30P aggregates were demonstrated to form in fly (Feany and Bender, Reference Feany and Bender2000) and rodent (Klein et al., Reference Klein, King, Hamby and Meyer2002; Lo Bianco et al., Reference Lo Bianco, Ridet, Schneider, Deglon and Aebischer2002; Lauwers et al., Reference Lauwers, Bequé, Van Laere, Nuyts, Bormans, Mortelmans, Casteels, Vercammen, Bockstael, Nuttin, Debyser and Baekelandt2007; da Silveira et al., Reference da Silveira, Schneider, Cifuentes-Diaz, Sage, Abbas-Terki, Iwatsubo, Unser and Aebischer2009) models. These differences may be related to the reduced affinity for lipid bilayers of this latter variant (Bodner et al., Reference Bodner, Maltsev, Dobson and Bax2010). It will be interesting to study these mechanisms in more detail and also to carry out studies in vivo of the more recently identified familial PD mutants E46K, H50Q and G51D, which appear to be less aggregation prone in vitro (Flagmeier et al., Reference Flagmeier, Meisl, Vendruscolo, Knowles, Dobson, Buell and Galvagnion2016), to shed more light on the aggregation process with respect to toxicity.
Oligomers have been suggested to play a key role in the toxicity of α-synuclein, although their molecular features in vivo have remained elusive. Stable α-synuclein oligomers can be generated in vitro and are toxic to neuronal cells in culture, causing increased levels of reactive oxygen species (ROS) (Cremades et al., Reference Cremades, Cohen, Deas, Abramov, Chen, Orte, Sandal, Clarke, Dunne, Aprile, Bertoncini, Wood, Knowles, Dobson and Klenerman2012; Chen et al., Reference Chen, Drakulic, Deas, Ouberai, Aprile, Arranz, Ness, Roodveldt, Guilliams, De-Genst, Klenerman, Wood, Knowles, Alfonso, Rivas, Abramov, Valpuesta, Dobson and Cremades2015). Detailed structural examination of stable toxic compared to non-toxic α-synuclein oligomers has revealed that toxicity is linked to strong binding to membrane surfaces and to insertion of a well-structured core into the lipid bilayer, whereas the more disordered species do not enter the bilayer and are not toxic (Fusco et al., Reference Fusco, Chen, Williamson, Cascella, Perni, Jarvis, Cecchi, Vendruscolo, Chiti, Cremades and Ying2017). This proposed mechanism of toxicity is interesting in the context of the interference with vesicle trafficking and lipid metabolism that was found in the in vivo models of α-synuclein as described in the Section ‘Biological pathways in Parkinson's disease models’, highlighting the power of connecting the observations made in cellular and animal models with detailed biophysical characterisation in vitro to understand the mechanisms behind protein misfolding diseases.
In vivo models of protein misfolding diseases
A common way to create an in vivo model of a protein misfolding disease is through the transgenic expression of a human gene associated with the disease in a non-human organism. The gene may typically encode for the protein that misfolds and aggregates, or for a protein that regulates this process. These transgenic models are often based on mutant genes known from familial cases of the disease in order to obtain a strong phenotype with high penetrance. Overexpression is usually employed to obtain high levels of the disease-associated protein, which facilitates the detection of a response in the organism.
Commonly used methods of probing the process of protein misfolding and aggregation in transgenic models include staining with amyloid-binding dyes or conformation-specific antibodies, ultrastructural examination by EM, and biochemical assays based on the solubility, the size or the shape of the misfolded species (Table 1). In parallel, the phenotype of the model can be assessed both at the organismal and the cellular levels, with the goal of correlating the conformational state of the disease-associated protein to neurotoxicity. In this section, we discuss this approach in the context of yeast, worm, fly and rodent models based on the transgenic expression of Aβ, tau or α-synuclein (Fig. 3).
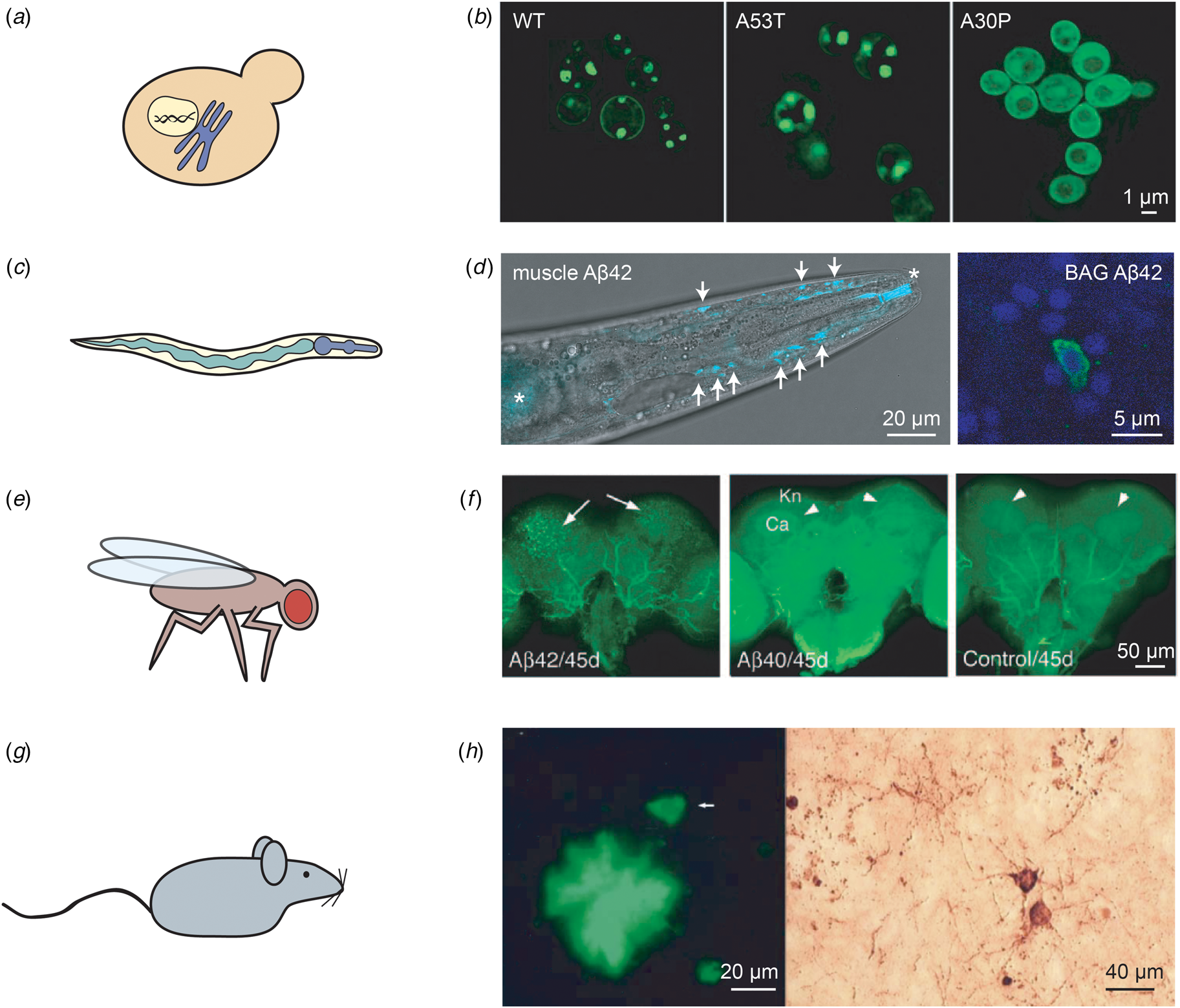
Fig. 3. Examples of in vivo models of protein misfolding for the study of AD and PD. (a) The budding yeast S. cerevisiae is the most widely used eukaryotic model organism, featuring a conserved cellular organisation, including the nucleus and the secretory system. (b) The expression of two copies of human α-synuclein (either in the wild-type or the familial A53T and A30P forms) in S. cerevisiae leads to the formation of cytoplasmic puncta for the wild-type and A53T variants, but not for A30P; reproduced from Outeiro and Lindquist (Reference Outeiro and Lindquist2003) with permission from Science. (c) The nematode worm C. elegans is a multicellular organism that consists of several tissues, such as a digestive system comprised of the pharynx and the intestine. (d) Left: C. elegans expressing Aβ42 in the body wall muscle cells display deposits that can be stained with the amyloid-binding dye X-34. The head region of an Aβ42-expressing animal is shown, with deposits indicated by arrows; asterisks mark unspecific staining of the mouth and intestine. Right: Close-up of C. elegans expressing Aβ42 in the two BAG neurons, stained with an anti-Aβ antibody (green) and DAPI (blue). Adapted from Sinnige et al. (Reference Sinnige, Ciryam, Casford, Dobson, De Bono and Vendruscolo2019). (e) The fruit fly D. melanogaster has a more complex brain than C. elegans, and possesses a compound eye that is often used as a read-out for toxicity. (f) The expression of Aβ42, but not of Aβ40, leads to the formation of ThS-positive deposits. The arrows indicate deposits; Kn: Kenyon cell layer; Ca: calyx. Reproduced from Iijima et al. (Reference Iijima, Liu, Chiang, Hearn, Konsolaki and Zhong2004); copyright 2004, National Academy of Sciences. (g) Rodent models are typically used for pre-clinical studies, but also provide a more complex system for fundamental studies of human disease mechanisms. (h) Overexpression in mice of human APP and tau with familial mutations leads to the formation of Aβ deposits positive for ThS staining (left) and structures resembling tau tangles as visualised by Bielschowsky silver staining (right). The arrow in the left panel indicates a neuron. Reproduced from Ribé et al. (Reference Ribé, Pérez, Puig, Gich, Lim, Cuadrado, Sesma, Catena, Sánchez, Nieto, Gómez-Ramos, Morán, Cabodevilla, Samaranch, Ortiz, Pérez, Ferrer, Avila and Gómez-Isla2005) with permission from Elsevier.
Aβ models
Yeast models
Yeast, in particular Saccharomyces cerevisiae, is one of the simplest and most-studied eukaryotic organisms available for the study of cellular pathologies (Verduyckt et al., Reference Verduyckt, Vignaud, Bynens, Van den Brande, Franssens, Cullin, Winderickx, Castrillo and Oliver2016; Tenreiro et al., Reference Tenreiro, Franssens, Winderickx and Outeiro2017; Seynnaeve et al., Reference Seynnaeve, Del Vecchio, Fruhmann, Verelst, Cools, Beckers, Mulvihill, Winderickx and Franssens2018; Rencus-Lazar et al., Reference Rencus-Lazar, DeRowe, Adsi, Gazit and Laor2019). This organism has a short generation time, and an arsenal of research tools is available for its study (Smith and Snyder, Reference Smith and Snyder2006). S. cerevisiae was the first eukaryote to have its complete genome sequenced (Goffeau et al., Reference Goffeau, Barrell, Bussey, Davis, Dujon, Feldmann, Galibert, Hoheisel, Jacq, Johnston, Louis, Mewes, Murakami, Philippsen, Tettelin and Oliver1996), and we now know that at least 60% of the S. cerevisiae genes have significant homology, or share conserved domains, with human genes (Khurana and Lindquist, Reference Khurana and Lindquist2010). However, as not all homologues have conserved functions, and not all human disease-related proteins have a yeast homologue, an often-used strategy to create a disease model is to express the human disease-related gene in yeast.
Yeast lacks known homologues of APP and β- and γ-secretases, yet transgenic variants of this organism have been successfully employed to investigate APP processing (Zhang et al., Reference Zhang, Komano, Fuller, Gandy and Frail1994, Reference Zhang, Espinoza, Hines, Innis, Mehta and Miller1997; Lüthi et al., Reference Lüthi, Schaerer-Brodbeck, Tanner, Middendorp, Edler and Barberis2003) and to screen for inhibitors of β-secretase (Middendorp et al., Reference Middendorp, Ortler, Neumann, Paganetti, Lüthi and Barberis2004) and Aβ-mediated toxicity (Bharadwaj et al., Reference Bharadwaj, Verdile, Barr, Gupta, Steele, Lenard Lachenmayer, Yue, Ehrlich, Petsko, Ju, Ringe, Sankovich, Caine, MacReadie, Gandy and Martins2012; Amen and Kaganovich, Reference Amen and Kaganovich2016; Seynnaeve et al., Reference Seynnaeve, Del Vecchio, Fruhmann, Verelst, Cools, Beckers, Mulvihill, Winderickx and Franssens2018). It has proven challenging, however, to generate yeast models to observe the full Aβ aggregation process and its associated toxicity (Verduyckt et al., Reference Verduyckt, Vignaud, Bynens, Van den Brande, Franssens, Cullin, Winderickx, Castrillo and Oliver2016; Seynnaeve et al., Reference Seynnaeve, Del Vecchio, Fruhmann, Verelst, Cools, Beckers, Mulvihill, Winderickx and Franssens2018). Expression of Aβ42 in fusion with green fluorescent protein (GFP-Aβ42) in this organism was found to result in small punctate patches in newly transformed cells, which become larger as the cells age, and a modest reduction of about 5% in cellular growth was observed (Caine et al., Reference Caine, Sankovich, Antony, Waddington, Macreadie, Varghese and Macreadie2007). Cells expressing an Aβ42 fusion protein with the MRF domain of the yeast prion Sup35p have been reported as a model system for Aβ42 oligomerisation, where a decrease in activity of the reporter was shown to be associated with the formation of sodium dodecyl sulphate (SDS)-stable oligomers (Bagriantsev and Liebman, Reference Bagriantsev and Liebman2006).
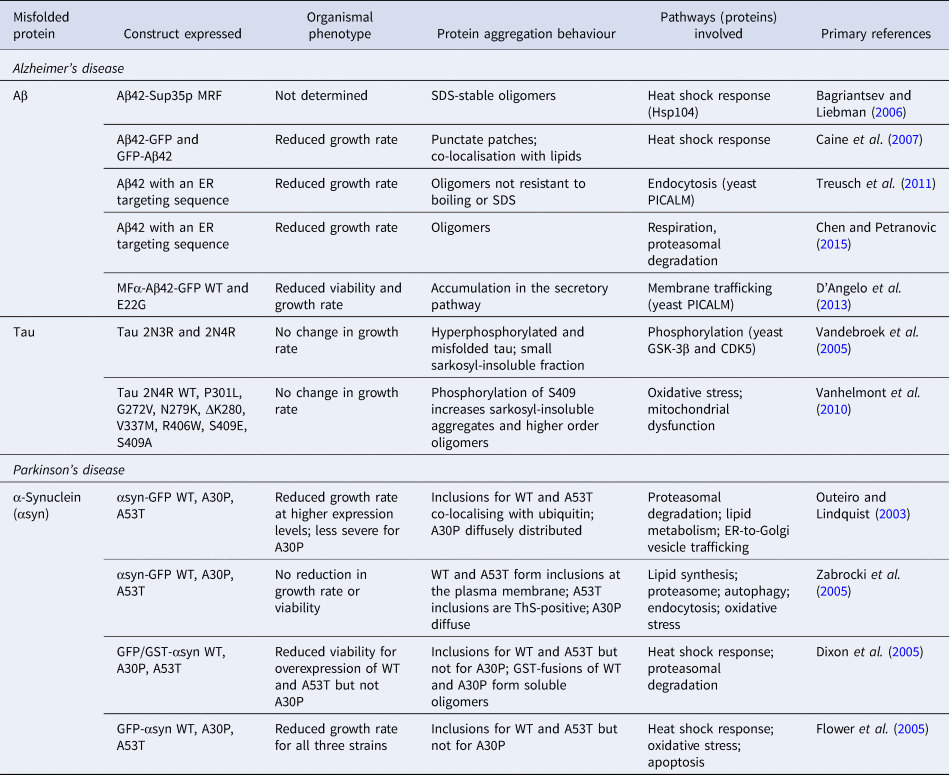
To model the generation of Aβ in the secretory pathway, experiments have been carried out using the peptide fused to an endoplasmic reticulum (ER)-targeting sequence (Treusch et al., Reference Treusch, Hamamichi, Goodman, Matlack, Chung, Baru, Shulman, Parrado, Bevis, Valastyan, Han, Lindhagen-persson, Reiman, Evans, Bennett, Olofsson, Dejager, Tanzi, Caldwell, Caldwell and Lindquist2011). This model was shown to lead to the accumulation of Aβ in the secretory pathway, accompanied by growth defects, which were more severe for Aβ42 than for Aβ40. Consistent with these observations, Aβ42 was shown to form more oligomers than Aβ40. These observations were confirmed in an independent study in which the expression of Aβ42 with the ER-targeting sequence was furthermore shown to lead to impaired mitochondrial function and proteasomal activity (Chen and Petranovic, Reference Chen and Petranovic2015). Further to these results, expression of an MFα-Aβ42-GFP fusion construct that also enters the secretory pathway and causes toxicity was found to be exacerbated for the familial, more aggregation-prone E22G Arctic mutant (D'Angelo et al., Reference D'Angelo, Vignaud, Di Martino, Salin, Devin, Cullin and Marchal2013), although the aggregation state of the fusion proteins was not examined. An overview of the various yeast models used in the study of Aβ aggregation is presented in Table 2.
Table 2. List of yeast models of protein misfolding in AD and PD
Worm models
The nematode worm C. elegans has been used extensively to investigate the molecular processes underlying the cytotoxicity of Aβ aggregation (Link, Reference Link2006; Alexander et al., Reference Alexander, Marfil and Li2014), and for the discovery of compounds capable of inhibiting such processes (Habchi et al., Reference Habchi, Chia, Limbocker, Mannini, Ahn, Perni, Hansson, Arosio, Kumita, Challa, Cohen, Linse, Dobson, Knowles and Vendruscolo2016; Perni et al., Reference Perni, Challa, Kirkegaard, Limbocker, Koopman, Hardenberg, Sormanni, Müller, Saar, Roode, Habchi, Vecchi, Fernando, Casford, Nollen, Vendruscolo, Dobson and Knowles2018). C. elegans has the advantage over yeast that it is multicellular, yet has a relatively simple body plan with 959 cells for the adult hermaphrodite, including a nervous system comprised of 302 neurons. The complete genome of C. elegans was sequenced in 1998 (The C. elegans Sequencing Consortium, 1998), and follow-up studies showed that 38% of the C. elegans genes have known human homologues (Shaye and Greenwald, Reference Shaye and Greenwald2011). The nematode typically lives up to 2–3 weeks under laboratory conditions, a convenient experimental timescale, and it has been shown to exhibit age-associated phenotypes, such as deterioration of muscle structure, as observed in higher organisms including humans (Herndon et al., Reference Herndon, Schmeissner, Dudaronek, Brown, Listner, Sakano, Paupard, Hall and Driscoll2002).
Although C. elegans contains the APP homologue apl-1, this gene lacks a sequence region homologous to human Aβ. Moreover, as C. elegans lacks the β-secretase activity required to process full-length APP, transgenic models have been largely based on the overexpression of the human Aβ42 sequence (Table 3). In the first reported model, Aβ42 was placed under control of the unc-54 promoter for expression in the body wall muscle cells (Link, Reference Link1995). For efficient expression, a signal sequence upstream of Aβ was found to be required, and the peptide was shown to be properly processed, yet Aβ deposits were found in the cytoplasm of the muscle cells rather than in the secretory system or in the extracellular space (Link et al., Reference Link, Johnson, Fonte, Paupard, Hall, Styren, Mathis and Klunk2001). However, co-immunoprecipitation has revealed that Aβ interacts with the ER-homologue of the Hsp70 chaperone in this model, suggesting that it enters the secretory pathway, but is then rerouted to the cytoplasm for degradation (Fonte et al., Reference Fonte, Kapulkin, Taft, Fluet, Friedman and Link2002). Examination using amyloid-specific dyes and immuno-EM has shown that a subset of the deposits is fibrillar, whereas a dimeric Aβ construct forms deposits but without fibrillar structures (Fay et al., Reference Fay, Fluet, Johnson and Link1998; Link et al., Reference Link, Johnson, Fonte, Paupard, Hall, Styren, Mathis and Klunk2001). From a set of mutants designed to interfere with β-strand formation, the L17P and M35C variants also fail to produce deposits that stain with amyloid-sensitive dye ThS (Fay et al., Reference Fay, Fluet, Johnson and Link1998).
Table 3. List of worm models of protein misfolding in AD and PD
Subsequently, it was found that in the original model Aβ is processed to residues 3-42 (McColl et al., Reference McColl, Roberts, Gunn, Perez, Tew, Masters, Barnham, Cherny and Bush2009), a fragment that is a major component of human amyloid plaques with the N-terminal residue modified to a pyroglutamate (Harigaya et al., Reference Harigaya, Saido, Eckman, Prada, Shoji and Younkin2000). A model has therefore been created with two extra residues in between the signal sequence and that of Aβ, resulting in expression of the 1-42 peptide (McColl et al., Reference McColl, Roberts, Pukala, Kenche, Roberts, Link, Ryan, Masters, Barnham, Bush and Cherny2012). In both strains, amyloidogenic protein aggregation (Fig. 3d, left panel) results in paralysis of the worms within several days of adulthood, yet the strain expressing Aβ(1-42) is more temperature sensitive and shows increased paralysis at higher temperatures, perhaps reflecting differences in the aggregation behaviour of the two isoforms (McColl et al., Reference McColl, Roberts, Pukala, Kenche, Roberts, Link, Ryan, Masters, Barnham, Bush and Cherny2012).
A model in which Aβ(3-42) is expressed in neurons features oligomeric species recognised by the NU-4 antibody, and displays a phenotype with impaired chemotaxis (Wu et al., Reference Wu, Wu, Butko, Christen, Lambert, Klein, Link and Luo2006) and defects in associative learning (Dosanjh et al., Reference Dosanjh, Brown, Rao, Link and Luo2010). Interestingly, these worms are defective in sensing benzaldehyde, but not diacetyl, suggesting that the vulnerability of the associated sensory pathways towards Aβ toxicity differs. In a more recent model expressing the corrected Aβ(1-42) variant throughout the nervous system, the protein was found to accumulate in the insoluble fraction during ageing, whereas neuromuscular defects and behavioural abnormalities were observed earlier on (Fong et al., Reference Fong, Teo, Ng, Chen, Lakshmanan, Tsoi, Moore, Inoue, Halliwell and Gruber2016). Expressing Aβ specifically in glutamatergic neurons results in visible neurodegeneration of the glutamatergic neurons in the tail of the worm (Treusch et al., Reference Treusch, Hamamichi, Goodman, Matlack, Chung, Baru, Shulman, Parrado, Bevis, Valastyan, Han, Lindhagen-persson, Reiman, Evans, Bennett, Olofsson, Dejager, Tanzi, Caldwell, Caldwell and Lindquist2011). However, a model designed in our own laboratory that selectively expresses low levels of Aβ42 in the two BAG sensory neurons, which are also glutamatergic, shows moderate behavioural defects in a CO2 sensing assay without overt signs of neurodegeneration, perhaps because these neurons are more resistant to toxic protein aggregation than others (Sinnige et al., Reference Sinnige, Ciryam, Casford, Dobson, De Bono and Vendruscolo2019). Notably, in this model Aβ42 appears to be localised mainly in the cell body (Fig. 3d, right panel) and does not visibly form aggregates, supporting this notion.
Fly models
Drosophila melanogaster, the fruit fly, is a common model organism for neurodegenerative diseases (Rincon-Limas et al., Reference Rincon-Limas, Jensen and Fernandez-Funez2012; Prüßing et al., Reference Prüßing, Voigt and Schulz2013) (Table 4), whose main advantage over C. elegans is its more sophisticated nervous system. The central nervous system of the fly comprises glia as well as neurons, and contains specialised sub-structures such as a compound eye and mushroom bodies for olfactory learning and memory, both of which are often employed in disease models. Limitations for the modelling of neurodegenerative disorders, on the other hand, are the absence of an adaptive immune system, and a lack of blood vessels.
Table 4. List of fruit fly models of protein misfolding in AD and PD
a All Aβ40/42 expressed using signal peptide.
Similar to C. elegans, Drosophila lacks a sequence region corresponding to human Aβ in its APP homologue. A β-secretase homologue does exist (Carmine-Simmen et al., Reference Carmine-Simmen, Proctor, Tschäpe, Poeck, Triphan, Strauss and Kretzschmar2009), but has low levels of activity (Fossgreen et al., Reference Fossgreen, Brückner, Czech, Masters, Beyreuther and Paro1998; Greeve et al., Reference Greeve, Kretzschmar, Tschape, Beyn, Brellinger, Schweizer, Nitsch and Reifegerste2004). γ-Secretase activity has also been demonstrated by expression in Drosophila of a construct comprising the Aβ region, the transmembrane region and the C-terminal segment of human APP, leading to the generation of a 4 kDa Aβ fragment that may correspond to either Aβ40 or Aβ42 (Fossgreen et al., Reference Fossgreen, Brückner, Czech, Masters, Beyreuther and Paro1998). A transgenic model for Aβ overproduction has been designed in which human APP is expressed together with human β-secretase in the eye, resulting in the generation of a 4 kDa Aβ peptide and progressive degeneration of the retina and neuropil regions (Greeve et al., Reference Greeve, Kretzschmar, Tschape, Beyn, Brellinger, Schweizer, Nitsch and Reifegerste2004). ThS-positive inclusions with a star-like appearance have been found in older flies, yet neurodegeneration precedes the formation of these amyloid deposits. Co-expression of human APP and β-secretase with Drosophila PSEN containing mutations corresponding to those in familial AD was found to reduce the onset of amyloid deposition remarkably, from 68 days to 37 days, and to cause more severe neurodegeneration (Greeve et al., Reference Greeve, Kretzschmar, Tschape, Beyn, Brellinger, Schweizer, Nitsch and Reifegerste2004).
Expression of human APP and β-secretase throughout the Drosophila nervous system was found to result in Aβ deposition in the cortical layer, as shown by antibody staining, and memory defects in young flies, followed by extensive neurodegeneration in older flies (Sarantseva et al., Reference Sarantseva, Timoshenko, Bolshakova, Karaseva, Rodin, Schwarzman and Vitek2009). Using a similar approach, a model has been generated that displays a more severe phenotype, including abnormal wing morphology at the time of eclosion and strong neuroanatomical changes, suggesting a developmental defect. In this model, punctate Aβ deposits, which stain with the amyloid-binding dye X-34, are present in relatively young flies (Chakraborty et al., Reference Chakraborty, Vepuri, Mhatre, Paddock, Miller, Michelson, Delvadia, Desai, Vinokur, Melicharek, Utreja, Khandelwal, Ansaloni, Goldstein, Moir, Lee, Tabb, Saunders and Marenda2011). The reason for the discrepancy between these two models has been suggested to lie in the different caloric intake of the flies, which may affect γ-secretase processing (Chakraborty et al., Reference Chakraborty, Vepuri, Mhatre, Paddock, Miller, Michelson, Delvadia, Desai, Vinokur, Melicharek, Utreja, Khandelwal, Ansaloni, Goldstein, Moir, Lee, Tabb, Saunders and Marenda2011). Raising the flies at lower temperatures was found to result in a milder phenotype, with Aβ deposits accumulating during ageing, accompanied by impaired climbing and memory functions and visible neurodegeneration (Mhatre et al., Reference Mhatre, Michelson, Gomes, Tabb, Saunders and Marenda2014).
To avoid the complications associated with APP processing, another approach has been exploited in which Aβ40 and Aβ42 are directly expressed following the pre-proenkephalin signal peptide, which was shown to be cleaved correctly (Finelli et al., Reference Finelli, Kelkar, Song, Yang and Konsolaki2004). Flies expressing Aβ42 have a rough eye phenotype and the peptide accumulates in the insoluble protein fraction, whereas flies expressing the less aggregation-prone Aβ40 appear normal (Finelli et al., Reference Finelli, Kelkar, Song, Yang and Konsolaki2004). The lines that were created have varying expression levels of Aβ42 in the eye, depending on the integration site of the construct and the number of copies. Aggregation and toxicity were found to be proportional to the expression levels, with low Aβ42 levels showing small deposits and modest neurodegeneration, and higher levels resulting in larger deposits and more severe morphological defects (Finelli et al., Reference Finelli, Kelkar, Song, Yang and Konsolaki2004).
Expression of Aβ42 throughout the nervous system leads to locomotory and other behavioural deficits in relatively young flies, in addition to a dramatically reduced lifespan (Finelli et al., Reference Finelli, Kelkar, Song, Yang and Konsolaki2004). Further characterisation of this model has revealed that Aβ42 forms deposits in various regions of the brain, a subset in the region of the Kenyon cell bodies being ThS positive (Fig. 3f) (Iijima et al., Reference Iijima, Liu, Chiang, Hearn, Konsolaki and Zhong2004). However, fibrils could not be identified by using EM, suggesting that Aβ42 forms diffuse, relatively unstructured deposits rather than mature amyloid plaques in this model. Similar experiments with Aβ40 have shown that it accumulates in the same brain areas, but deposits are not visible. Although both Aβ40 and Aβ42 expression cause age-associated learning defects, only Aβ42 results in locomotory defects and neurodegeneration at later ages. This observation suggests that visible protein aggregation is required in this model to observe neurodegeneration, but not neuronal dysfunction, which could perhaps be mediated by oligomers (Iijima et al., Reference Iijima, Liu, Chiang, Hearn, Konsolaki and Zhong2004).
Using the signal peptide of the Drosophila necrotic gene, lines have been generated expressing Aβ40 and Aβ42, and also Aβ42 with the Arctic mutation E22G in the Drosophila nervous system (Crowther et al., Reference Crowther, Kinghorn, Miranda, Page, Curry, Duthie, Gubb and Lomas2005). All Aβ variants were found initially to accumulate in intracellular deposits, which react with an antibody specific to oligomeric Aβ (Crowther et al., Reference Crowther, Kinghorn, Miranda, Page, Curry, Duthie, Gubb and Lomas2005). In older flies, Aβ42 shows extracellular deposits accompanied by vacuolar structures, which are exacerbated in the E22G mutant, whereas they do not occur in Aβ40 flies. The extracellular deposits do not stain with Congo red, indicating that they do not consist of mature amyloid fibrils, but might resemble diffuse plaques as in the previously described fly model (Iijima et al., Reference Iijima, Liu, Chiang, Hearn, Konsolaki and Zhong2004). The Aβ42 flies have a rough eye phenotype and locomotory defects, which occur before the appearance of the extracellular deposits, and the phenotype is exacerbated by the Arctic mutation. Furthermore, the Aβ42 flies have a shorter lifespan than controls, whereas the survival of the Aβ40 flies is indistinguishable from controls. The phenotype of the Aβ42 flies can be partially restored by treatment with the amyloid-specific dye Congo red, suggesting that toxicity is directly caused by Aβ42 aggregation.
In a subsequent study, a panel of Aβ42 mutants with different aggregation propensities has been expressed in the Drosophila nervous system. The computationally predicted aggregation rates were found to correlate strongly with lifespan and locomotory function, especially when the propensity to form protofibrillar (oligomeric) aggregates was considered (Luheshi et al., Reference Luheshi, Tartaglia, Brorsson, Pawar, Watson, Chiti, Vendruscolo, Lomas, Dobson and Crowther2007). Similarly, whereas expression of Aβ40 itself is relatively harmless to the flies, point mutations that increase the formation of prefibrillar Aβ40 species in vitro result in a toxic phenotype when expressed in Drosophila (Brorsson et al., Reference Brorsson, Bolognesi, Tartaglia, Shammas, Favrin, Watson, Lomas, Chiti, Vendruscolo, Dobson, Crowther and Luheshi2010). Furthermore, an engineered affibody has been shown to restore the phenotype of Aβ42 flies, by binding to the monomer and targeting it for degradation (Luheshi et al., Reference Luheshi, Hoyer, de Barros, van Härd, Brorsson, Macao, Persson, Crowther, Lomas, Ståhl, Dobson and Härd2010). On the other hand, the KW1 antibody, which is specific to Aβ40 oligomers, induces aggregation into toxic protofibrillar species, shortening the lifespan of the flies (Wacker et al., Reference Wacker, Rönicke, Westermann, Wulff, Reymann, Dobson, Horn, Crowther, Luheshi and Fändrich2014). Further evidence for the role of intermediate species in toxicity has been provided by the expression of tandem constructs of Aβ40 and Aβ42, in which two monomers are connected with a linker to increase the effective concentrations and aggregation rates. Expression of both tandem constructs leads to the formation of deposits in the fly brain, but only the tandem Aβ42 flies show a dramatic reduction in lifespan relative to those expressing the monomeric peptide, an effect that is associated with an increase in SDS-soluble species (Speretta et al., Reference Speretta, Jahn, Tartaglia, Favrin, Barros, Imarisio, Lomas, Luheshi, Crowther and Dobson2012).
Aβ42, the E22G Arctic mutant, as well as a designed mutant with lower aggregation propensity have furthermore been expressed in the Drosophila nervous system using the pre-proenkephalin signal peptide (Iijima et al., Reference Iijima, Chiang, Hearn, Hakker, Gatt, Shenton, Granger, Leung, Iijima-Ando and Zhong2008). In this study, the Aβ variants were found to be localised intracellularly, residing in the ER, Golgi and lysosomes, but in contrast to the model by Crowther and co-workers, no oligomer-specific antibody staining was observed. None of the lines was found to show Congo red staining, demonstrating that mature fibrils are not noticeably formed either. The Arctic mutant shows an increased population of dimers and trimers by western blot, a larger number of ThS-positive deposits in the cell bodies accompanied by neuronal loss, a shortened lifespan and stronger locomotory defects than the model expressing wild-type Aβ42. The designed mutant, however, forms more ThS-positive deposits in the neurites leading to neuropil loss, and has a longer lifespan, yet shows learning defects similar to the Arctic mutant. These discrepancies have been suggested to arise from a differential intracellular distribution of the variants, in addition to their different aggregation propensities, highlighting the importance of the biological environment in mediating cellular toxicity (Iijima et al., Reference Iijima, Chiang, Hearn, Hakker, Gatt, Shenton, Granger, Leung, Iijima-Ando and Zhong2008).
Further studies on flies expressing Aβ variants in the giant fibre neurons have demonstrated that intracellular Aβ, which accumulates in cell bodies and axons, leads to impairments in synaptic function, accompanied by reduced numbers of mitochondria and synaptic vesicles (Zhao et al., Reference Zhao, Wang, Tan, Huang, Zhang, Zhang, Wang, YangCheng, Zhu, Sun and Huang2010). Again, the severity of the phenotype was found to be correlated with expression levels and to be stronger for the Arctic mutant. In addition, it has been shown that levels of the synaptic protein bruchpilot and presynaptic calcium channels decline with age in flies expressing the Arctic mutant, compared to controls (Huang et al., Reference Huang, Ma, Ji, Zhao, Tan, Sun and De Huang2013). This Drosophila model expressing Aβ in a subset of neurons has thus proved to be suitable to study perturbations at the single-neuron level, and has provided strong evidence for a connection between Aβ accumulation and synaptic dysfunction even in this relatively simple model organism.
Rodent models
One of first-transgenic mouse models of AD shown to develop amyloid plaques resembling those in the human disease relied on the overexpression of the V717F mutant of human APP, which is associated with early-onset AD, driven by the platelet-derived growth factor-β (PDGF-β) promoter (Games et al., Reference Games, Adams, Alessandrini, Barbour, Berthelette, Blackwell, Carr, Clemens, Donaldson and Gillespie1995). These mice, referred to as PDAPP from the combination of PDGF-β and APP, show ThS-positive extracellular plaques accompanied by dystrophic neurites and reactive astrocytes. The plaques vary in morphology from round to diffuse. The pathology is age-related, with plaques first appearing between 6 and 9 months of age, and increasing in number as the mice age further. This model displays synaptic loss correlated with increased plaque deposition in older mice (Dodart et al., Reference Dodart, Mathis, Saura, Bales, Paul and Ungerer2000) as well as learning defects (Chen et al., Reference Chen, Chen, Knox, Inglis, Bernard, Martin, Justice, McConlogue, Games, Freedman and Morris2000), but without overt neuronal loss (Irizarry et al., Reference Irizarry, Soriano, McNamara, Page, Schenk, Games and Hyman1997).
A wide variety of models based on mutant APP overexpression has been subsequently generated, which similarly display amyloid plaques and are associated with synaptic dysfunction and gliosis (reviewed in Elder et al. (Reference Elder, Gama Sosa and De Gasperi2010); Hall and Roberson (Reference Hall and Roberson2012); Drummond and Wisniewski (Reference Drummond and Wisniewski2017); see http://www.alzforum.org for a full list of available rodent models and Table 5 for those discussed here). These models differ from one another in various ways including the ratio of Aβ40 to Aβ42, the morphology of the plaques, the age at which they appear and the brain regions that are affected. A widely used model is the Tg2576 mouse, in which human APP is overexpressed with the Swedish double mutation K670N/M671L using the prion protein (PrP) promoter that drives strong expression in neurons (Hsiao et al., Reference Hsiao, Chapman, Nilsen, Eckman, Harigaya, Younkin, Yang and Cole1996). In this model, dense ThS-positive deposits are found in older mice (11–13 months), a subset of which displays Congo red birefringence. Dystrophic neurites have also been found in this case, as well as reactive astrocytes and microglia. At the age where plaques are apparent, the mice show behavioural impairment in the Morris water maze test. However, there is no direct correlation between the quantity of plaques and cognitive defects, as the behavioural phenotype remains stable between 6 and 14 months, while the plaque load continues to increase. Rather, the levels of a 56 kDa soluble Aβ species, referred to as Aβ*56, identified in extracts from Tg2576 mice by SDS-PAGE and size exclusion chromatography (SEC) (Table 1) correlate with memory impairment (Lesné et al., Reference Lesné, Ming, Kotilinek, Kayed, Glabe, Yang, Gallagher and Ashe2006). Furthermore, this species affects long-term memory when purified and infused into young rats, supporting the hypothesis that Aβ oligomers are responsible for neurotoxicity in AD.
Table 5. List of rodent models of protein misfolding in AD and PD
A complication of APP-based transgenic mouse models is that its processing gives rise to several variants of the Aβ peptide, as well as other intracellular and extracellular fragments of APP (Fig. 2a), which may all contribute to the observed phenotype. To address specifically the role of Aβ accumulation, a different approach has been taken in expressing constructs comprising the BRI protein fused to either Aβ40 or Aβ42, which are released by the furin cleavage site directly preceding the Aβ sequence (McGowan et al., Reference McGowan, Pickford, Kim, Onstead, Eriksen, Yu, Skipper, Murphy, Beard, Das, Jansen, DeLucia, Lin, Dolios, Wang, Eckmann, Dickson, Hutton, Hardy and Golde2005). Expression of the Aβ40 construct leads to high levels of free peptide, which however remains soluble, so that no amyloid plaques are observed. In contrast, BRI-Aβ42 mice show amyloid pathology starting at 3 months of age, including the presence of diffuse and compact ThS-positive plaques, as well as deposits in blood vessels in the brain, termed cerebral amyloid angiopathy. However, although dystrophic neurites and reactive astrogliosis have been observed surrounding the plaques, this study reported that the mice have no behavioural abnormalities, suggesting that Aβ42 deposition by itself is not sufficient to trigger neurotoxicity, at least up to the age investigated (McGowan et al., Reference McGowan, Pickford, Kim, Onstead, Eriksen, Yu, Skipper, Murphy, Beard, Das, Jansen, DeLucia, Lin, Dolios, Wang, Eckmann, Dickson, Hutton, Hardy and Golde2005). It is interesting to compare these findings to those from the Drosophila and C. elegans models discussed earlier, where Aβ fusions do lead to behavioural phenotypes. These observations can perhaps be attributed to toxicity arising from intracellular rather than extracellular Aβ in these worm and fly models. Intracellular Aβ may also be present in APP-based mouse models (LaFerla et al., Reference LaFerla, Green and Oddo2007), but not in those developed using the BRI fusion approach.
Mutations in the PSEN genes PSEN1 and PSEN2 associated with early onset forms of AD have been employed to increase the levels of the more aggregation-prone Aβ42 in mouse models. Expression of familial mutant, but not wild-type, PSEN1 has been shown to increase specifically the levels of Aβ42 over Aβ40 in mice (Borchelt et al., Reference Borchelt, Thinakaran, Eckman, Lee, Davenport, Ratovitsky, Prada, Kim, Seekins, Yager, Slunt, Wang, Seeger, Levey, Gandy, Copeland, Jenkins, Price, Younkin and Sisodia1996; Duff et al., Reference Duff, Eckman, Zehr, Yu, Prada, Perez-Tur, Hutton, Buee, Harigaya, Yager, Morgan, Gordon, Holcomb, Refolo, Zenk, Hardy and Younkin1996; Citron et al., Reference Citron, Westaway, Xia, Carlson, Diehl, Levasque, Johnson-Wood, Lee, Seubert, Davis, Kholodenko, Motter, Sherrington, Perry, Yao, Strome, Lieberburg, Rommens, Kim, Schenk, Fraser, St George Hyslop and Selkoe1997), and crossing these lines with APP mutant mice has led to accelerated Aβ deposition (Borchelt et al., Reference Borchelt, Ratovitski, Van Lare, Lee, Gonzales, Jenkins, Copeland, Price and Sisodia1997; Holcomb et al., Reference Holcomb1998). Several PSAPP mouse models based on different combinations of APP and PSEN1/2 mutants exist, and all show similar pathologies to that of the APP-transgenic mice, but at an earlier age and accompanied by a stronger behavioural phenotype. They also suffer from greater neuronal loss, although not at levels comparable to the widespread degeneration in human AD (McGowan et al., Reference McGowan, Eriksen and Hutton2006; Elder et al., Reference Elder, Gama Sosa and De Gasperi2010; Hall and Roberson, Reference Hall and Roberson2012). For example, the PS2APP mouse model, a cross of the APP K670N/M671L mutant and PSEN2 with the N141I mutation, displays congophilic amyloid plaques at 9 months of age, which are surrounded by dystrophic neurites and activated microglia and astrocytes (Richards et al., Reference Richards, Higgins, Ouagazzal, Ozmen, Kew, Bohrmann, Malherbe, Brockhaus, Loetscher, Czech, Huber, Bluethmann, Jacobsen and Kemp2003). However, deficiencies in synaptic plasticity and behavioural tests such as the Morris water maze are relatively mild (Richards et al., Reference Richards, Higgins, Ouagazzal, Ozmen, Kew, Bohrmann, Malherbe, Brockhaus, Loetscher, Czech, Huber, Bluethmann, Jacobsen and Kemp2003).
In the APPSLPS1KI mouse model, which expresses APP with V717I and K670N/M671L mutations, and PSEN1 with M233T and L235P mutations, large numbers of plaques are already apparent at 6 months of age, and about 50% neuronal loss in the CA1/2 layer of the hippocampus has been observed at 10 months of age (Casas et al., Reference Casas, Sergeant, Itier, Blanchard, Wirths, van der Kolk, Vingtdeux, van de Steeg, Ret, Canton, Drobecq, Clark, Bonici, Delacourte, Benavides, Schmitz, Tremp, Bayer, Benoit and Pradier2004). The loss of neurons appears to correlate better with the presence of intracellular Aβ aggregates that can be stained with ThS than with extracellular plaques, which is interesting given evidence for the presence of intracellular Aβ in human AD (LaFerla et al., Reference LaFerla, Green and Oddo2007). Coinciding with this pathology, the mice become increasingly impaired in working memory and motor tasks from 6 months of age (Wirths et al., Reference Wirths, Breyhan, Schäfer, Roth and Bayer2008). Apart from the fact that this model produces Aβ42 as the dominant Aβ species, another interesting feature in relation to human AD is the progressive N-terminal truncation and modification of Aβ42 into species that have been detected in patient material (Casas et al., Reference Casas, Sergeant, Itier, Blanchard, Wirths, van der Kolk, Vingtdeux, van de Steeg, Ret, Canton, Drobecq, Clark, Bonici, Delacourte, Benavides, Schmitz, Tremp, Bayer, Benoit and Pradier2004).
A model with very extensive amyloid deposition has been created using the L166P mutant form of PSEN1, which has led to a disease onset as young as 24 years of age in humans, crossed with a mouse strain expressing APP K670N/M671L (Radde et al., Reference Radde, Bolmont, Kaeser, Coomaraswamy, Lindau, Stoltze, Calhoun, Jäggi, Wolburg, Gengler, Haass, Ghetti, Czech, Hölscher, Mathews and Jucker2006). In the resulting model, congophilic amyloid plaques appear in the neocortex at 6 weeks, whereas deposits in different regions of the hippocampus occur between 2 and 5 months. As in the other mouse models, the plaques are surrounded by dystrophic neurites and glia, but cognitive impairment is only evident at 8 months (Radde et al., Reference Radde, Bolmont, Kaeser, Coomaraswamy, Lindau, Stoltze, Calhoun, Jäggi, Wolburg, Gengler, Haass, Ghetti, Czech, Hölscher, Mathews and Jucker2006).
The approach of combining APP and PSEN mutants has been further extended through the generation of a mouse strain with a total of five familial AD mutations, the 5xFAD mouse, which has become one of the most frequently used models in AD research (Oakley et al., Reference Oakley, Cole, Logan, Maus, Shao, Craft, Guillozet-Bongaarts, Ohno, Disterhoft, Van Eldik, Berry and Vassar2006). Constructs of APP with K670N/M671L, I716V and V717I mutations, and PSEN1 with M146L/L286V, were co-injected to create this model, resulting in the rapid accumulation of Aβ42 and ThS-positive plaques from 2 months of age. The plaques are smaller but more numerous than in the Tg2576 mouse, consistent with an increase in nucleation events due to the higher concentration of Aβ42. In this model, intracellular Aβ aggregation also appears to play a role, as indicated by small ThS-positive puncta in pyramidal neurons. It has been speculated that extracellular plaques could arise from these aggregates upon cell death, as indeed degenerating neurons with intracellular deposits are sometimes found in the vicinity of plaques. These regions of co-localisation are correlated with synaptic degeneration and significant loss of neurons. At the behavioural level, there is no phenotype at 2 months in these mice, but at 4–5 months, memory defects are apparent in the Y-maze test (Oakley et al., Reference Oakley, Cole, Logan, Maus, Shao, Craft, Guillozet-Bongaarts, Ohno, Disterhoft, Van Eldik, Berry and Vassar2006).
Mouse models based on the overexpression of human genes produce artificially high protein levels, including other APP fragments in addition to Aβ, altogether complicating the interpretation of the phenotype. Second-generation mouse models remedy some of these issues by making use of knock-in strategies to produce human Aβ in the context of endogenous mouse APP (Sasaguri et al., Reference Sasaguri, Nilsson, Hashimoto, Nagata, Saito, De Strooper, Hardy, Vassar, Winblad and Saido2017). For example, the Aβ sequence of mouse APP was humanised, and Swedish (K670M/M671L) and Iberian (I716F) mutations were introduced in order to generate high levels of preferentially Aβ42 (Saito et al., Reference Saito, Matsuba, Mihira, Takano, Nilsson, Itohara, Iwata and Saido2014). The resulting APPNL-F mice display progressive Aβ pathology and inflammation, accompanied by memory impairment in the Y-maze test. Adding the Arctic mutation E693G has resulted in the APPNL-G-F model that displays earlier Aβ deposition and a stronger inflammatory response.
Another strategy to avoid overexpression consists in triggering an amyloid cascade response by intracerebral infusion of Aβ seed aggregates produced either in vitro or derived from cell systems, other animal models or AD patients (Jucker and Walker, Reference Jucker and Walker2018). These non-transgenic mouse models are particularly useful in the study of the mechanisms of propagation of the aggregates across cells and brain tissues, but the infusion process tends to generate inflammatory reactions that complicate the interpretation of the results.
Given that rats are evolutionarily more closely related to humans than mice, and display more complex patterns of behaviour, rat models have certain advantages for recapitulating and testing the pathogenic mechanisms behind AD. Although early rat models transgenic for APP did not display amyloid plaques, probably due to insufficient levels of expression (reviewed in Do Carmo and Cuello, Reference Do Carmo and Cuello2013), the McGill-R-Thy1-APP model has been successful in generating many aspects of AD pathology with just a single transgene of APP combining Swedish (K670N/M671L) and Indiana (V717F) mutations (Leon et al., Reference Leon, Canneva, Partridge, Allard, Ferretti, Dewilde, Vercauteren, Atifeh, Ducatenzeiler, Klein, Szyf, Alhonen and Cuello2010). These rats accumulate intracellular Aβ from just 1 week after birth, and the first amyloid deposits show at 6 months in a particular brain region, the subiculum. Investigations in older animals have specifically identified degenerating glutamatergic and cholinergic neurons surrounding compact, ThS-positive plaques, showing the vulnerability of these types of neurons. Furthermore, the levels of soluble trimeric Aβ species are greatly elevated in homozygous rats, and were found to correlate with learning and memory deficits at 3 months of age (Leon et al., Reference Leon, Canneva, Partridge, Allard, Ferretti, Dewilde, Vercauteren, Atifeh, Ducatenzeiler, Klein, Szyf, Alhonen and Cuello2010). This observation points towards toxicity of soluble Aβ species, rather than mature amyloid plaques. This model thus recapitulates many features of human AD, although a subsequent study has shown that neuronal loss is in fact very limited, being largely restricted to the subiculum at 18 months of age (Heggland et al., Reference Heggland, Storkaas, Soligard, Kobro‐Flatmoen and Witter2015).
Another useful rat model, TgF344-AD, has been developed by expressing K670N/M671L mutant APP and PSEN1 with a deletion of exon 9 (Cohen et al., Reference Cohen, Rezai-Zadeh, Weitz, Rentsendorj, Gate, Spivak, Bholat, Vasilevko, Glabe, Breunig, Rakic, Davtyan, Agadjanyan, Kepe, Barrio, Bannykh, Szekely, Pechnick and Town2013a). At 16 months of age, the rats display widespread deposition of ThS-positive amyloid plaques in the cingulate cortex and hippocampus, as well as the striatum and cerebellum, all of which are regions affected by amyloid deposition in AD patients. The model also features intraneuronal accumulation of Aβ42, and soluble oligomeric Aβ species have been found which correlate with significant neuronal loss; in addition the model displays inflammation and phagocytosis of neuronal debris. At the behavioural level, the rats show defects in various assays, and these progress with age following the development of the observed pathology. In contrast to most APP/PSEN-based mouse models, this rat model displays tau pathology consisting of Gallyas silver-stained tangles both in vicinity of the amyloid plaques, and also in regions devoid of plaques, as observed in human AD (Braak and Braak, Reference Braak and Braak1991). The presence of tau tangles is critical to study the interplay between Aβ and tau pathology, which is still poorly understood, as outlined in the next sections.
Tau models
Yeast models
Yeast models have been employed to compare the in vivo aggregation propensities of various tau isoforms and disease-associated mutations, as well as the role of specific phosphorylation sites (Table 2). Upon expression of the human tau isoforms 2N3R and 2N4R in yeast, a small fraction of both protein constructs accumulates in the sarkosyl-insoluble aggregate fraction, with the 2N4R isoform being more extensively phosphorylated than the 2N3R isoform (Vandebroek et al., Reference Vandebroek, Vanhelmont, Terwel, Borghgraef, Lemaire, Snauwaert, Wera, Van Leuven and Winderickx2005). Comparison of a set of six familial point mutants in the 2N4R isoform showed that all become hyperphosphorylated, and accumulate more strongly in the sarkosyl-insoluble fraction than the wild-type protein (Vanhelmont et al., Reference Vanhelmont, Vandebroek, De Vos, Terwel, Lemaire, Anandhakumar, Franssens, Swinnen, Van Leuven and Winderickx2010). Purified hyperphosphorylated, soluble wild-type tau quickly forms structured filaments in vitro, and is able to seed tau isolated from transgenic yeast cells (Vandebroek et al., Reference Vandebroek, Vanhelmont, Terwel, Borghgraef, Lemaire, Snauwaert, Wera, Van Leuven and Winderickx2005). The P301L tau variant found in FTDP-17 patients, purified from yeast, has been shown to aggregate in vitro when presented with a microtubule surface as a substrate, in contrast to wild-type tau-4R (Vandebroek et al., Reference Vandebroek, Terwel, Vanhelmont, Gysemans, Van Haesendonck, Engelborghs, Winderickx and Van Leuven2006). Although yeast models have thus confirmed key aspects of tau phosphorylation and aggregation, no toxicity has been observed in any of the reported expression systems as judged from the growth rates of the cells (Vandebroek et al., Reference Vandebroek, Vanhelmont, Terwel, Borghgraef, Lemaire, Snauwaert, Wera, Van Leuven and Winderickx2005; De Vos et al., Reference De Vos, Anandhakumar, Van den Brande, Verduyckt, Franssens, Winderickx and Swinnen2011).
Worm models
The first C. elegans models of tauopathy have been based on the overexpression of a construct comprising human tau 1N4R in nerve cells (Kraemer et al., Reference Kraemer, Zhang, Leverenz, Thomas, Trojanowski and Schellenberg2003). In addition to that expressing the wild-type protein, lines expressing the mutants P301L and V337M, which are associated with frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP-17), have been generated, all resulting in an uncoordinated phenotype with reduced thrashing rates and shortened lifespan. Tau becomes phosphorylated at the majority of known sites in these models, and it accumulates in the detergent-soluble fraction as the worms age, as well as in the detergent-insoluble fraction in the case of the mutant lines. In addition to presynaptic defects in cholinergic transmission, degeneration of GABAergic axons has been observed. However, tau was found to be diffusely localised in the axons, and only the V337M variant showed clear deposits in electron micrographs. This mutant line contains the highest level of insoluble tau, and displays the strongest phenotype, but the results obtained on wild-type and P301L tau suggest that the formation of deposits is not required for neurodegeneration (Kraemer et al., Reference Kraemer, Zhang, Leverenz, Thomas, Trojanowski and Schellenberg2003).
A study in which the 3R and 4R isoforms of wild-type tau, and the 4R isoforms of P301L and R406W were expressed specifically in the six mechanosensory neurons of C. elegans has largely corroborated these findings (Miyasaka et al., Reference Miyasaka, Ding, Gengyo-Ando, Oue, Yamaguchi, Mitani and Ihara2005). The touch response is only mildly affected in worms expressing wild-type tau, but declines strongly with age in the mutant strains, both of which show corresponding signs of neurodegeneration. Ultrastructural examination by using EM has shown accumulation of tau in the cell bodies and neuronal processes with concomitant loss of microtubules, yet no evidence for fibrillar aggregates was found.
Expression of different tau variants with increased or decreased aggregation propensities has, however, highlighted the relevance of toxic protein aggregates in the C. elegans models. Co-expression of V337M tau with a short aggregation-prone tau fragment, F3ΔK280, leads to the formation of ThS-positive inclusions, and increased accumulation of full-length tau in the detergent-soluble and detergent-insoluble fractions during ageing compared to the strain expressing V337M tau alone (Fatouros et al., Reference Fatouros, Pir, Biernat, Koushika, Mandelkow, Mandelkow, Schmidt and Baumeister2012). Concomitantly, the locomotory phenotype is more severe, the neurons display more morphological abnormalities, and the transport of mitochondria is perturbed. The introduction of mutations predicted to prevent the formation of β-strands in the F3ΔK280 fragment has the opposite effect, and in addition compounds known to inhibit tau aggregation have been shown to reduce the accumulation of insoluble tau and partially restore the locomotory phenotype of the strain expressing the aggregation-prone fragment (Fatouros et al., Reference Fatouros, Pir, Biernat, Koushika, Mandelkow, Mandelkow, Schmidt and Baumeister2012).
Fly models
The first observation of tau toxicity in flies was serendipitous, when a reporter construct consisting of Drosophila tau tagged with GFP was expressed in sensory neurons with the aim to study axonal development. Morphological defects were observed, providing the first demonstration of tau toxicity in the fly, and similar results were obtained upon the expression of human tau (Williams et al., Reference Williams, Tyrer and Shepherd2000). Since then, Drosophila has been used extensively to study tau aggregation and toxicity, as well as the effects of phosphorylation by different kinases (see Sun and Chen (Reference Sun and Chen2014) for a more complete overview, and Table 4 for the models here discussed). As in yeast and C. elegans, the effects of FTDP-17 mutations have also been examined, for example in models expressing human wild-type and R406W tau forms throughout the nervous system (Wittmann et al., Reference Wittmann, Wszolek, Shulman, Salvaterra, Lewis, Hutton and Feany2001). Although both of these strains display a reduction in lifespan, the phenotype is stronger for the R406W mutant, and severe neurodegeneration is evident from vacuolar structures in the cortex. Both the wild-type and R406W tau forms are recognised by antibodies specific to pathogenic phosphorylation and misfolded conformations of tau, but evidence for fibrillar structures has not been found in investigations by EM.
Co-expression of the Drosophila homologue of the kinase GSK-3β with full-length tau in the fly eye does result in the characteristic flame-shaped aggregates resembling those found in human tauopathies (Jackson et al., Reference Jackson, Wiedau-Pazos, Sang, Wagle, Brown, Massachi and Geschwind2002). The antibody AT100, which is specific for phosphorylated tau in tangles, recognises tau only when co-expressed with GSK-3β in the fly. Co-expression of the kinase results in fibrillar aggregates with the dimensions of human paired helical filaments (PHFs) and straight filaments (SFs) as observed by EM, and exacerbates neurodegeneration compared to the expression of tau alone (Jackson et al., Reference Jackson, Wiedau-Pazos, Sang, Wagle, Brown, Massachi and Geschwind2002). In agreement with the toxic effects of phosphorylation by GSK-3β, inhibition of the kinase restores the locomotory phenotype in a model expressing human wild-type tau 0N3R in motor neurons (Mudher et al., Reference Mudher, Shepherd, Newman, Mildren, Jukes, Squire, Mears, Berg, MacKay, Asuni, Bhat and Lovestone2004). Another kinase, PAR-1, has been shown to phosphorylate tau upstream of GSK-3β, and its overexpression exacerbates the rough eye phenotype of the R406W model, but in the absence of visible tau aggregates (Nishimura et al., Reference Nishimura, Yang and Lu2004).
In certain tauopathies, fibrillary tangles occur in glia in addition to neurons (Lee et al., Reference Lee, Goedert and Trojanowski2001). A Drosophila model in which human 0N4R tau is expressed specifically in adult glia shows a reduction in lifespan and the presence of apoptotic signals in glia and, by non-cell autonomous mechanisms, in cholinergic neurons (Colodner and Feany, Reference Colodner and Feany2010). In aged flies, tau is found in sarkosyl-insoluble fractions, which contain straight fibrils as shown by EM studies. Immunohistochemistry has confirmed that the glia contain inclusions consisting of phosphorylated tau, which are furthermore positive for ThS and Bielschowsky silver staining, and contain fibrillar structures in EM. Expression of tau in both glia and neurons leads to increased toxicity compared to that in neurons alone, as measured by the number of apoptotic cells, indicating the importance of glial tau in these neurodegenerative disorders.
In a recent study, a comparison of a panel of Drosophila models expressing wild-type tau and five FTDP-17-linked mutations at similar expression levels has further strengthened the correlation between tau phosphorylation and neurotoxicity (Bardai et al., Reference Bardai, Wang, Mutreja, Yenjerla, Gamblin and Feany2018). Mutants that display the strongest aggregation, indicated by the number of inclusions in vivo and ThS fluorescence of the corresponding recombinant proteins in vitro, show the least caspase activation and induction of stress signalling, demonstrating that mature aggregates do not cause significant toxicity. The mutants generating the highest toxicity, on the other hand, have the highest phosphorylation levels and show increased F-actin accumulation, suggesting a correlation between actin stabilisation and toxicity (Bardai et al., Reference Bardai, Wang, Mutreja, Yenjerla, Gamblin and Feany2018).
Rodent models
As in the case of APP-based mouse models, initial rodent models overexpressing human wild-type tau constructs have not displayed clear behavioural phenotypes, nor produced significant numbers of tau tangles, which is why most models have been generated using tau with mutations found in FTDP-17 (reviewed in Hutton et al., Reference Hutton, Lewis, Dickson, Yen and McGowan2001; Lee et al., Reference Lee, Kenyon and Trojanowski2005). An overview of about 30 current transgenic mouse models of tau is available at http://www.alzforum.org; we highlight here those that are commonly used, or that have led to particularly interesting insights (Table 5).
One of the few successful models based on wild-type tau is the htau model, a cross of a strain expressing human tau, including all regulatory regions and leading to all splicing variants, with a knock-out strain for endogenous mouse tau (Andorfer et al., Reference Andorfer, Kress, Espinoza, De Silva, Tucker, Barde, Duff and Davies2003). Expression of the human construct by itself does not lead to clear tau pathology (Duff et al., Reference Duff, Knight, Refolo, Sanders, Yu, Picciano, Malester, Hutton, Adamson, Goedert, Burki and Davies2000), suggesting that the interplay between mouse (which only occurs as 4R) and human isoforms is important. In the htau model, tau was observed to be correctly localised in the axons in 6-week-old animals, but tau staining was found to shift towards the cell bodies as the animals age, accompanied by increased phosphorylation at particular sites. Beyond 9 months, tau aggregation was inferred from immunohistochemistry and immuno-EM, and the sarkosyl-insoluble fraction was found to contain PHFs with similar dimensions to the ones found in AD brains (Andorfer et al., Reference Andorfer, Kress, Espinoza, De Silva, Tucker, Barde, Duff and Davies2003). The mice suffer significant neuronal loss at old age, between 8 and 18 months, but the majority of dying neurons does not appear to contain fibrillar aggregates (Andorfer et al., Reference Andorfer, Acker, Kress, Hof, Duff and Davies2005). However, memory impairments and synaptic defects occur at 12 months, but not at 4 months, indicating that tau pathology does need to develop to a certain stage to cause damage (Polydoro et al., Reference Polydoro, Acker, Duff, Castillo and Davies2009).
A more recent model based on wild-type tau, Tau4RTg2652, has been generated to overexpress high levels of 1N4R wild-type tau, driven by the Thy1.2 promoter, resulting in approximately 12-fold overexpression relative to endogenous mouse tau (Wheeler et al., Reference Wheeler, McMillan, Hawk, Iba, Robinson, Xu, Dombroski, Jeong, Dichter, Juul, Loomis, Raskind, Leverenz, Trojanowski, Lee, Schellenberg and Kraemer2015). In this model, misfolded and phosphorylated tau is detectable by various antibodies in the forebrain, amygdala, hippocampus and brainstem even at 3 months, but tangles are not observed by ThS and Gallyas staining even at 24 months of age. The levels of oligomeric tau as recognised by the TOC1 antibody are relatively low, but abnormal axon morphologies and dystrophic neurites have been nevertheless identified throughout the brain. The mice show a slight motor phenotype, but the most obvious behavioural phenotype is an impairment in spatial memory as assessed in the Barnes maze test. Altogether, these data suggest that pre-tangle tau, consisting of altered conformations and phosphorylation states, but not oligomers or fibrils, could be sufficient to drive neuronal dysfunction. Both the tau pathology and the behavioural phenotype remain stable from 3 months onwards, which may make this model suitable for rapid studies of the early stages of sporadic tauopathies, including AD.
Most other existing tau models are based on tau mutations linked to FTDP-17, which are not directly related to AD, but provide strong phenotypes, which are useful for fundamental studies of tau pathology, as well as for preclinical trials targeted at halting tau toxicity. The JNPL3 mouse, overexpressing tau 0N4R with the P301L mutation from the PrP promoter, shows impaired motor function and abundant tau tangles in several regions of the brain as well as the spinal cord; the latter are, however, typically not associated with AD (Lewis et al., Reference Lewis, McGowan, Rockwood, Melrose, Nacharaju, Van Slegtenhorst, Gwinn-Hardy, Paul Murphy, Baker, Yu, Duff, Hardy, Corral, Lin, Yen, Dickson, Davies and Hutton2000). The tau accumulations are recognised by a range of conformational and phospho-specific antibodies, and are positive for Congo red, ThS and other staining agents such as Gallyas silver. Different morphologies have been found, including the flame-shaped tangles characteristic of AD, but also aggregates resembling Pick bodies and small, dense inclusions similar to those found in FTDP-17. EM studies have confirmed the presence of fibrils, and sarkosyl-insoluble protein has been found in extracts of brain and spinal cord from aged JNPL3 mice, but not from control lines expressing wild-type human tau.
In another mouse model, known as pR5, the P301L mutant is expressed in the context of the longest tau isoform 2N4R under the control of the Thy1.2 promoter (Götz et al., Reference Götz, Chen, van Dorpe and Nitsch2001a). In this model, tau also accumulates in various brain regions, and forms tangles stained by ThS and Gallyas silver in e.g. the spinal cord and layers 5 and 6 of the cortex. Reactive astrocytes and apoptotic neurons have been found in the same regions. Other neurons, including pyramidal cells in the hippocampus, show granular tau accumulation that is recognised by a subset of phospho- and conformation-specific antibodies. The sarkosyl-insoluble fraction of brain extracts contains fibrils as observed by using EM, but they are shorter and narrower than fibrils obtained from AD patient material. This is in line with the observation that P301L fibrils from FTDP-17 patients have a different morphology from that of V337M FTDP-17 fibrils or wild-type AD fibrils (Spillantini et al., Reference Spillantini, Crowther, Kamphorst, Heutink and van Swieten1998b), suggesting that the point mutations can significantly affect the fibril structure. Furthermore, the strong overexpression in mouse models may give rise to shorter fibrils than those observed in AD, because the likelihood of primary nucleation is higher.
The rTg4510 model has also been designed to express P301L tau (0N4R), in this case using an elegant expression system that can be inhibited with doxycycline (Ramsden et al., Reference Ramsden, Kotilinek, Forster, Paulson, McGowan, SantaCruz, Guimaraes, Yue, Lewis, Carlson, Hutton and Ashe2005; SantaCruz et al., Reference SantaCruz, Lewis, Spires, Paulson, Kotilinek, Ingelsson, Guimaraes, DeTure, Ramsden, McGowan, Forster, Yue, Orne, Janus, Mariash, Kushowski, Hyman, Hutton and Ashe2005). The Ca2+/calmodulin kinase II (CaMKII) promoter has been used to drive expression specifically in the forebrain, where the first tau tangles appear at 2.5 months of age, leading up to extensive tau pathology and increasing atrophy in the cortex and hippocampus from 5 months onwards (Ramsden et al., Reference Ramsden, Kotilinek, Forster, Paulson, McGowan, SantaCruz, Guimaraes, Yue, Lewis, Carlson, Hutton and Ashe2005). Misfolded tau conformations and phosphorylation also develop with age, as indicated using various antibodies, and the main tau species shifts from the soluble 55 kDa form at 2.5 months to the sarkosyl-insoluble, extensively phosphorylated 64 kDa species at 8.5 months. At the ultrastructural level, SFs have been observed using EM on fixed tissue samples from 10-month-old animals, which occasionally adopt a typical herring bone pattern which has also been observed in deposits in Pick's disease. At the behavioural level, spatial memory in the Morris water maze test is impaired from 4 months of age, and locomotory deficits have been observed in 10-month old mice (Ramsden et al., Reference Ramsden, Kotilinek, Forster, Paulson, McGowan, SantaCruz, Guimaraes, Yue, Lewis, Carlson, Hutton and Ashe2005).
The possibility of lowering tau expression in this model has led to interesting insights into the relationship between tau pathology and toxicity (SantaCruz et al., Reference SantaCruz, Lewis, Spires, Paulson, Kotilinek, Ingelsson, Guimaraes, DeTure, Ramsden, McGowan, Forster, Yue, Orne, Janus, Mariash, Kushowski, Hyman, Hutton and Ashe2005). Suppressing tau expression at 2.5 months causes the pathology to be halted. In contrast, when doxycycline treatment is given at 4 months or later, tau phosphorylation and accumulation in the insoluble fraction proceed, and numbers of tau tangles grow similarly as in untreated animals. Given the significant reduction in tau mRNA, this suggests that the soluble species that were already present can develop into pathological tau. However, brain atrophy and memory defects were found to be largely restored if the doxycycline treatment was sufficiently long, demonstrating that the presence of tau tangles is not directly correlated with the disruption of cognitive function (SantaCruz et al., Reference SantaCruz, Lewis, Spires, Paulson, Kotilinek, Ingelsson, Guimaraes, DeTure, Ramsden, McGowan, Forster, Yue, Orne, Janus, Mariash, Kushowski, Hyman, Hutton and Ashe2005).
A model expressing tau 1N4R with the P301S mutation, PS19, shows a strong motor phenotype which presents difficulties for behavioural studies, yet it has been a useful model because of its robust tau pathology and evident neurodegeneration (Yoshiyama et al., Reference Yoshiyama, Higuchi, Zhang, Huang, Iwata, Saido, Maeda, Suhara, Trojanowski and Lee2007). Phosphorylated tau has been shown to accumulate rapidly, and increased levels of insoluble tau have been observed during ageing. At 6 months of age, flame-shaped tau tangles and globular aggregates, as well as thread-like structures, can be visualised in the hippocampus, amygdala and spinal cord by Gallyas silver staining, and by staining with ThS and Congo red. Activated microglia and astrocytes, as well as neuronal inflammatory markers, develop from 3 months onwards. Tau pathology and inflammation precede large scale neurodegeneration, which is observed from 9 months onwards in the hippocampus and other brain regions. However, hippocampal synapses are already lost by 3 months of age, preceding the appearance of tau tangles. As such, the results obtained from this model add to the notion that neurodegeneration is initiated before mature protein aggregates are formed.
The ΔK280 mutation, which is known from a FTDP-17 case, is directly correlated with the aggregation propensity of tau, being located in a segment of the protein that forms a β-strand in the fibril structure. Mouse models based on this pro-aggregation mutant have been generated in parallel with an anti-aggregation mutant consisting of ΔK280 combined with two proline mutations (I277P and I308P), which strongly inhibit β-strand formation (Eckermann et al., Reference Eckermann, Mocanu, Khlistunova, Biernat, Nissen, Hofmann, Schönig, Bujard, Haemisch, Mandelkow, Zhou, Rune and Mandelkow2007; Mocanu et al., Reference Mocanu, Nissen, Eckermann, Khlistunova, Biernat, Drexler, Petrova, Schonig, Bujard, Mandelkow, Zhou, Rune and Mandelkow2008). These mutants have been expressed in the context of full-length human tau 2N4R (Eckermann et al., Reference Eckermann, Mocanu, Khlistunova, Biernat, Nissen, Hofmann, Schönig, Bujard, Haemisch, Mandelkow, Zhou, Rune and Mandelkow2007), and in a short construct comprising only the repeat domain of tau (Mocanu et al., Reference Mocanu, Nissen, Eckermann, Khlistunova, Biernat, Drexler, Petrova, Schonig, Bujard, Mandelkow, Zhou, Rune and Mandelkow2008) in two respective studies, leading to very similar results. The pro-aggregation mutant adopts a misfolded conformation that is recognised by the MC1 antibody, becomes phosphorylated at several sites and is partially found in the sarkosyl-insoluble fraction, whereas the anti-aggregation mutant does not display these features. Furthermore, the pro-aggregation mutant has been shown to induce co-aggregation of endogenous mouse tau, which co-localises with the exogenous human tau in the same filaments extracted from the mouse brain. In both studies, quantification from electron micrographs has shown that the pro-aggregation mutant causes more synaptic loss than the anti-aggregation mutant. The strong correlation between in vitro and in vivo aggregation propensities is furthermore underscored by the fact that the truncated construct leads to Gallyas-positive tau tangles much earlier than full-length tau, in line with a protective effect of the flanking regions on tau aggregation observed in vitro. These models also make use of the regulatable expression system, and switching off tau expression has been shown largely to reverse tau pathology (Eckermann et al., Reference Eckermann, Mocanu, Khlistunova, Biernat, Nissen, Hofmann, Schönig, Bujard, Haemisch, Mandelkow, Zhou, Rune and Mandelkow2007; Mocanu et al., Reference Mocanu, Nissen, Eckermann, Khlistunova, Biernat, Drexler, Petrova, Schonig, Bujard, Mandelkow, Zhou, Rune and Mandelkow2008), in contrast to the earlier study which reported that tau tangles continue to develop after reducing transgene expression, although memory function recovered (SantaCruz et al., Reference SantaCruz, Lewis, Spires, Paulson, Kotilinek, Ingelsson, Guimaraes, DeTure, Ramsden, McGowan, Forster, Yue, Orne, Janus, Mariash, Kushowski, Hyman, Hutton and Ashe2005). However, phosphorylation and aggregation of endogenous mouse tau persist in the pro-aggregation model, possibly seeded by the pathological human tau expressed prior to switching off the transgene (Mocanu et al., Reference Mocanu, Nissen, Eckermann, Khlistunova, Biernat, Drexler, Petrova, Schonig, Bujard, Mandelkow, Zhou, Rune and Mandelkow2008).
More recently, mouse models have been generated expressing tau with the A152T mutation, which has been found to increase the risk of FTD as well as AD. In one model, the mutation has been introduced in the context of full length tau 2N4R (Decker et al., Reference Decker, Krüger, Sydow, Dennissen, Siskova, Eckhard and Eva-Maria2016; Sydow et al., Reference Sydow, Hochgräfe, Könen, Cadinu, Matenia, Petrova, Joseph, Dennissen and Mandelkow2016), whereas another model that was simultaneously reported is based on the expression of tau 1N4R (Maeda et al., Reference Maeda, Djukic, Taneja, Yu, Lo, Davis, Craft, Guo, Wang, Kim, Ponnusamy, Gill, Masliah and Mucke2016). Both models display reactive microglia and astrocytes and neuronal hyperexcitability, with deficits in behavioural assays and neuronal loss occurring at later age. The model expressing A152T tau 2N4R was shown to develop ThS and Gallyas silver positive tau tangles starting from 3 months of age (Sydow et al., Reference Sydow, Hochgräfe, Könen, Cadinu, Matenia, Petrova, Joseph, Dennissen and Mandelkow2016), but in the model expressing the 1N4R isoform, the misfolded tau was found to remain largely soluble (Maeda et al., Reference Maeda, Djukic, Taneja, Yu, Lo, Davis, Craft, Guo, Wang, Kim, Ponnusamy, Gill, Masliah and Mucke2016). Thus, soluble A152T tau oligomers may be responsible for the overlapping phenotypes of these two models.
Rat-transgenic models have been used less extensively than mouse to study human tauopathies. A rat model expressing full-length tau with the P301L mutation displays tau phosphorylation and accumulation in the sarkosyl-insoluble fraction, but no tangles, nor behavioural abnormalities (Korhonen et al., Reference Korhonen, van Groen, Thornell, Kyrylenko, Soininen, Ojala, Peltomaa, Tanila, Salminen, Mandelkow and Soininen2011). Models based on truncated 4R and 3R isoforms (residues 151–391), which are thought to play a role in AD, do display full tau pathology including the presence of phosphorylated, misfolded and sarkosyl-insoluble tau, and ThS and Congo red positive tangles, but without overt neuronal loss (Zilka et al., Reference Zilka, Filipcik, Koson, Fialova, Skrabana, Zilkova, Rolkova, Kontsekova and Novak2006; Koson et al., Reference Koson, Zilka, Kovac, Kovacech, Korenova, Filipcik and Novak2008; Filipcik et al., Reference Filipcik, Zilka, Bugos, Kucerak, Koson, Novak and Novak2012).
Models combining Aβ and tau
Worm models
The link between Aβ and tau pathologies in AD has remained incompletely understood, in part because animal models have not been particularly successful in recapitulating both features of the disease. Crossing C. elegans strains expressing Aβ42 and an aggregation-prone tau mutant in neurons was shown to lead to an exacerbation of the toxic phenotype in terms of lifespan, viability of offspring, chemotaxis and other behavioural assays (Wang et al., Reference Wang, Saar, Leung, Chen and Wong2018). Conversely, crossing an anti-aggregation tau variant and an Aβ42 strain has not led to a significantly different phenotype compared to Aβ42 alone. The total aggregation load as observed from Congo red staining was found to be much increased in the double-transgenic line with the pro-aggregation tau mutant, and signs of neurodegeneration and neuronal loss were observed, but a mechanistic link between Aβ42 and tau toxicity has not been revealed from these studies.
Fly models
Co-expression of Aβ42 has been shown to exacerbate the phenotype of wild-type human tau-transgenic flies, manifesting in disrupted axonal transport, abnormal axonal morphology, impaired motor function and shortened lifespan (Folwell et al., Reference Folwell, Cowan, Ubhi, Shiabh, Newman, Shepherd and Mudher2010). This effect has been suggested to be mediated by increased tau phosphorylation in the presence of Aβ42, but the responsible kinases have so far not been identified. In an inducible fly model expressing Aβ42 with the Arctic mutation (E22G), the Drosophila kinase GSK-3β is activated and Aβ42-mediated toxicity is reduced when endogenous fly tau is removed (Sofola et al., Reference Sofola, Kerr, Rogers, Killick, Augustin, Gandy, Allen, Hardy, Lovestone and Partridge2010). However, differences in the phosphorylation of Drosophila tau in the presence or absence of Arctic Aβ42 are below detection limits. Thus, the link between Aβ42 and tau phosphorylation remains unclear from the Drosophila studies.
Rodent models
APP/PSEN-based rodent models have largely failed to induce significant tau pathology, although tau knock-out models have demonstrated that the presence of tau is required for Aβ-related toxicity to occur fully (Roberson et al., Reference Roberson, Scearce-Levie, Palop, Yan, Chen, Wu, Gerstein, Yu and Mucke2007; Leroy et al., Reference Leroy, Ando, Laporte, Dedecker, Suain, Authelet, Héraud, Pierrot, Yilmaz, Octave and Brion2012). To induce both Aβ and tau pathology, however, it has been necessary to develop double- or triple-transgenic models overproducing combinations of human APP, PSEN1/2 and tau, often with mutations that do not occur simultaneously in AD patients. Nevertheless, these models have proved to be useful for understanding the interactions between Aβ and tau pathology in AD.
The first-transgenic mouse model combining APP with tau has been the TAPP mouse, resulting from a cross of Tg2576, which expresses APP containing the Swedish mutation (K670N/M671L) with the JNPL3 mouse, which expresses the P301L tau variant (Lewis et al., Reference Lewis, Dickson, Lin, Chisholm, Corral, Jones, Yen, Sahara, Skipper, Yager, Eckman, Hardy, Hutton and Mcgowan2001). The number of tau tangles is highly increased in the TAPP mouse compared to the original JNPL3 mouse, and tangles are found in brain regions that are unaffected in the JNPL3 model, including the hippocampus, amygdala and entorhinal cortex. Other regions that are affected by tau pathology in the JNPL3 mouse, such as the hindbrain and spinal cord, do not show an increase. Aβ deposition is, however, not changed compared to the Tg2576 model, in line with the amyloid cascade hypothesis, according to which Aβ aggregation occurs upstream of tau pathology (Selkoe and Hardy, Reference Selkoe and Hardy2016). Despite the clear link, tau tangles are not identified in close vicinity of amyloid plaques, indicating that other mechanisms, perhaps mediated by intracellular Aβ, may play a role. These results have been confirmed by an independent study in which Aβ42 fibrils were injected into the pR5 mouse model which also expresses tau P301L, resulting in a 5-fold increase in tau tangles (Götz et al., Reference Götz, Chen, van Dorpe and Nitsch2001a, Reference Götz, Chen, Barmettler and Nitsch2001b). In this experiment, the main increase in tau tangles was found in neurons in the amygdala, which are distant but projecting towards the injection site, raising the possibility that retrograde transport of Aβ42 can promote tau tangle formation.
A triple-transgenic mouse model, the 3xTg-AD mouse, has been created by co-injecting constructs of the Swedish APP variant (K670N/M671L) and the P301L tau variant into embryos carrying the M146V PSEN1 variant (Oddo et al., Reference Oddo, Caccamo, Shepherd, Murphy, Golde, Kayed, Metherate, Mattson, Akbari and LaFerla2003). This model has been used extensively in AD research because the temporal and spatial appearance of amyloid plaques and tau tangles resembles that observed in the human disease. Antibody staining for Aβ is first observed intracellularly at 3–4 months of age, mostly in the neocortex and subsequently in the hippocampus. Plaques develop progressively in the same regions between 6 and 12 months. Tau pathology is initiated later and follows a different spatial pattern, first accumulating in pyramidal neurons in the hippocampus, followed by cortical regions. Aggregated tau can be recognised by conformation-specific and phospho-specific antibodies, as well as Gallyas silver and ThS staining. Although this sequence of events does not prove causality between Aβ and tau aggregation, both proteins have been found to co-localise within the same neurons, suggesting a role for intracellular Aβ in triggering tau pathology. Synaptic defects are observed in the hippocampus at 6 months in this model, which correlates with the presence of intracellular Aβ.
Models based on the PS19 line, expressing the P301S tau variant, have largely confirmed these findings. Crossing this line with the PDAPP mouse or the 5xFAD line has resulted in exacerbated tau pathology, whereas Aβ deposition is not significantly affected (Hurtado et al., Reference Hurtado, Molina-Porcel, Iba, Aboagye, Paul, Trojanowski and Lee2010; Saul et al., Reference Saul, Sprenger, Bayer and Wirths2013). The 5xFAD/PS19 model furthermore shows increased astrogliosis compared to the original transgenic lines, as well as increased loss of synapses and neuronal death (Saul et al., Reference Saul, Sprenger, Bayer and Wirths2013). The PDAPP/PS19 mice have been used for detailed analysis of the spatial progression of tau pathology, which matches to some extent the Braak staging of AD (Hurtado et al., Reference Hurtado, Molina-Porcel, Iba, Aboagye, Paul, Trojanowski and Lee2010). However, a limitation of the PS19 model and crosses thereof is their strong motor phenotype, which interferes with commonly used behavioural assays.
Another triple-transgenic model, TauPS2APP, has resulted from crossing the PS2APP line expressing the K670N/M671L APP variant and the N141I PSEN2, with the pR5 tau P301L strain (Grueninger et al., Reference Grueninger, Bohrmann, Czech, Ballard, Frey, Weidensteiner, von Kienlin and Ozmen2010). In contrast to the models described above, amyloid plaques and accumulations of phosphorylated tau develop in parallel in this model, and in the same regions of the hippocampus. Gallyas silver staining and immuno-EM confirmed that the deposits of tau are of fibrillar nature, and that structures resembling the PHFs observed in AD are present within dendrites. Phosphorylation of S422 has been shown by means of highly specific antibodies to be significantly increased in the triple-transgenic line, compared to the original tau model. However, the triple-transgenic mice do not show signs of increased neuronal loss compared to wild-type controls, and perform similarly to the PS2APP strain in behavioural assays.
PLB1 double mice have been generated by targeted knock-in of the APP K670N/M671L/V717I and tau P301L/R406W mutants (Platt et al., Reference Platt, Drever, Koss, Stoppelkamp, Jyoti, Plano, Utan, Merrick, Ryan, Melis, Wan, Mingarelli, Porcu, Scrocchi, Welch and Riedel2011), resulting in more moderate expression levels than in models generated by pronuclear injection, a process that leads to integration of multiple copies of the transgene. This line has been subsequently crossed with a PSEN1 A246E strain (Borchelt et al., Reference Borchelt, Ratovitski, Van Lare, Lee, Gonzales, Jenkins, Copeland, Price and Sisodia1997) to generate the PLB1 triple mouse model (Platt et al., Reference Platt, Drever, Koss, Stoppelkamp, Jyoti, Plano, Utan, Merrick, Ryan, Melis, Wan, Mingarelli, Porcu, Scrocchi, Welch and Riedel2011). Despite the low levels of Aβ in the triple strain, intracellular staining as well as a small number of extracellular ThS and Congo red positive plaques appear at 6 months of age in the hippocampus and cortex, and the number of plaques increases during ageing. Phosphorylated tau has been identified in the same brain regions from 6 months of age, but no evidence for mature tau tangles was found. Synaptic plasticity, together with social recognition and spatial memory, start to decline at the same age. Furthermore, glucose metabolism and sleep behaviour are altered in the PLB1 triple mice, as found in patients suffering from AD. Altogether, despite some discrepancies such as the relatively fast accumulation of phosphorylated tau, this less aggressive model recapitulates many features of the early phases of human AD.
Although most studies using double- and triple-transgenic mice have shown a unidirectional relationship between APP/Aβ expression and increased tau pathology, there is an example in which amyloid deposition is increased by mutant tau expression. In that work, Tg2576 expressing APP with the K670N/M671L Swedish mutation was crossed with a mutant tau line, tauVLW, containing G272V, P301L and R406W mutations (Fig. 3h) (Pérez et al., Reference Pérez, Ribe, Rubio, Lim, Morán, Ramos, Ferrer, Isla and Avila2005; Ribé et al., Reference Ribé, Pérez, Puig, Gich, Lim, Cuadrado, Sesma, Catena, Sánchez, Nieto, Gómez-Ramos, Morán, Cabodevilla, Samaranch, Ortiz, Pérez, Ferrer, Avila and Gómez-Isla2005). Not only does the expression of APPSwe lead to increased tau phosphorylation and accumulation in the sarkosyl-insoluble fraction (Pérez et al., Reference Pérez, Ribe, Rubio, Lim, Morán, Ramos, Ferrer, Isla and Avila2005), but expression of the tau mutant also increases the numbers of amyloid plaques by up to 5-fold at 16 months of age (Ribé et al., Reference Ribé, Pérez, Puig, Gich, Lim, Cuadrado, Sesma, Catena, Sánchez, Nieto, Gómez-Ramos, Morán, Cabodevilla, Samaranch, Ortiz, Pérez, Ferrer, Avila and Gómez-Isla2005). The double-transgenic animals also suffer from more pronounced neuronal loss than either APPSwe or tauVLW littermates (Ribé et al., Reference Ribé, Pérez, Puig, Gich, Lim, Cuadrado, Sesma, Catena, Sánchez, Nieto, Gómez-Ramos, Morán, Cabodevilla, Samaranch, Ortiz, Pérez, Ferrer, Avila and Gómez-Isla2005).
The models discussed so far have been based on tau mutations found in FTDP-17 patients, or combinations thereof, and they typically display tau pathology in the single-transgenic lines, albeit increased by expression of Aβ. In a model combining wild-type human tau 0N3R with APP K670N/M671L/V717I and PSEN1 M146L, tau tangles are absent (Boutajangout et al., Reference Boutajangout, Authelet, Blanchard, Touchet, Tremp, Pradier and Brion2004). However, in addition to APP-stained and ubiquitin-stained dystrophic neurites, tau-positive dystrophic neurites are also found surrounding amyloid plaques in these triple-transgenic mice. In these neurites, tau is recognised by a subset of phospho-specific and conformation-specific antibodies, but is not fibrillar, as indicated from a lack of Gallyas or Congo red staining. Similar observations have been reported for the TAPP and 3xTg-AD mice, although in those models tau tangles do occur in more distant regions from the Aβ plaques (Lewis et al., Reference Lewis, Dickson, Lin, Chisholm, Corral, Jones, Yen, Sahara, Skipper, Yager, Eckman, Hardy, Hutton and Mcgowan2001; Oddo et al., Reference Oddo, Caccamo, Shepherd, Murphy, Golde, Kayed, Metherate, Mattson, Akbari and LaFerla2003). Similarly, in a more recent model resulting from a cross of WT human tau 4R with APPswe and PSEN1 Δexon9, no evidence was found for the formation of tau tangles, although the mice were only examined up to 10 months of age (Jackson et al., Reference Jackson, Rudinskiy, Herrmann, Croft, Kim, Petrova, Ramos-Rodriguez, Pitstick, Wegmann, Garcia-Alloza, Carlson, Hyman and Spires-Jones2016). This model, however, displays increased plaque counts and dystrophic neurites compared to the APP/PSEN1 mice, although no increase in neuronal loss or astrogliosis was observed.
In contrast, another model based on human wild-type tau does feature the development of tangles. This model is the result of a cross between animals expressing APP ΔE693 (the Osaka mutant) and a tau construct comprising 3R and 4R isoforms (Umeda et al., Reference Umeda, Maekawa, Kimura, Takashima, Tomiyama and Mori2014). APP ΔE693 is not associated with amyloid plaque formation, but generates high levels of Aβ oligomers. These oligomers, detected by the 11A1 conformational antibody, appear earlier in double-transgenic mice than in the original APP line, being evident intracellularly in the regions of the hippocampus and cerebral cortex at 6 months of age. Furthermore, hyperphosphorylated tau, recognised by AT8 and PHF1 antibodies, is formed more rapidly in the CA3 region of the hippocampus and in the cortex in double-transgenic animals, appearing between 6 and 12 months. Tau tangles are present in the same regions as detected by Gallyas silver staining and EM from 18 months of age onwards. Loss of synapses and neuronal death in the CA3 region are also increased in the double-transgenic line, as are the memory deficits observed in the Morris water maze test. Altogether, whereas other models have failed to produce the full pathology of wild-type tau isoforms, the combination of tau with APP ΔE693 has had better success in doing so. This finding demonstrates that the presence of Aβ plaques is not a prerequisite for tau pathology and neuronal dysfunction to occur, but Aβ oligomers may be responsible for triggering this cascade instead.
Taking advantage of the knock-in strategy described in the Section ‘Aβ models’ for the APP-based models, a recent study reports on the humanisation of the mouse MAPT gene (Saito et al., Reference Saito, Mihira, Matsuba, Sasaguri, Hashimoto, Narasimhan, Zhang, Murayama, Higuchi, Lee, Trojanowski and Saido2019). Although the subcellular distribution of human tau appears normal in this model, crossing it with the APP knock-in model, APPNL−G−F, results in enhanced tau phosphorylation at several sites. Tau tangles were not identified, yet these more physiological models may prove useful for further studies into the relationship between Aβ and tau pathology.
Finally, it is worth noting that the spreading of tau pathology by means of neuronal connectivity has received much attention in recent years (Goedert et al., Reference Goedert, Eisenberg and Crowther2017; Jucker and Walker, Reference Jucker and Walker2018). In a model expressing the human P301L tau variant exclusively in layer II of the entorhinal cortex, it has been shown that this protein is present in synaptically connected neurons that do not express it, and that it seeds the aggregation of endogenous mouse tau (de Calignon et al., Reference de Calignon, Polydoro, Suárez-Calvet, William, Adamowicz, Kopeikina, Pitstick, Sahara, Ashe, Carlson, Spires-Jones and Hyman2012). Crossing this line with the model that expresses APPswe and PSEN1 Δexon9 has led to an increased spread of misfolded tau, and to increased neuronal loss in the entorhinal cortex where tau is expressed (Pooler et al., Reference Pooler, Polydoro, Maury, Nicholls, Reddy, Wegmann, William, Saqran, Cagsal-Getkin, Pitstick, Beier, Carlson, Spires-Jones and Hyman2015). In addition, at 16 months of age, the triple-transgenic animals display more and larger Aβ deposits in the entorhinal and somatosensory cortices, and more dystrophic neurites surrounding the plaques.
In summary, although it has proved to be difficult to recapitulate the complex pathology and phenotype of human AD in rodent models, there has been significant progress towards this objective as a result of combining in a variety of ways transgenes for APP, PSENs and tau.
α-Synuclein models
Yeast models
The first yeast model for PD, based on the expression of human α-synuclein in S. cerevisiae, was reported in 2003 (Fig. 3b) (Outeiro and Lindquist, Reference Outeiro and Lindquist2003). Expression of a single copy of GFP-tagged human α-synuclein resulted in the localisation of the protein at lipid membranes, whereas expression of two copies caused it to form cytoplasmic inclusions containing lipid molecules, as shown by staining with the lipophilic dye Nile red, in addition to protein. Furthermore, a dose-dependent cytotoxicity was observed, consistent with a toxic gain of function associated with α-synuclein expression. Two familial α-synuclein mutants A30P and A53T were also investigated in this study. While the A53T mutant was found to display very similar behaviour to wild-type α-synuclein, the A30P mutant was shown to maintain a cytoplasmic distribution at single copy level, rather than accumulating at the lipid membrane, and to exhibit toxicity only when expressed from a high-copy plasmid (Outeiro and Lindquist, Reference Outeiro and Lindquist2003). Subsequently, inclusions of A53T α-synuclein, either GFP-tagged or unlabelled, were shown to react with ThS, indicating an amyloid-like structure, whereas staining was not observed in cells expressing the A30P mutant (Zabrocki et al., Reference Zabrocki, Pellens, Vanhelmont, Vandebroek, Griffioen, Wera, Van Leuven and Winderickx2005). These in vivo models (Table 2) have confirmed the importance of the interactions of α-synuclein with lipid membranes, and have provided insights into the correlation between inclusion formation, amyloid-like aggregation and cytotoxicity.
Worm models
The tendency of α-synuclein to accumulate into cytoplasmic inclusions was further corroborated in C. elegans models (Table 3). A model overexpressing yellow-fluorescent protein (YFP)-tagged α-synuclein in the body wall muscle cells displays inclusions from day 1 of adulthood, and these increase in number as the worms age (van Ham et al., Reference van Ham, Thijssen, Breitling, Hofstra, Plasterk and Nollen2008). Interestingly, fluorescence recovery after photobleaching (FRAP) measurements (Table 1) have indicated that α-synuclein remains mobile within the inclusions, and only in old worms (from day 11 onwards) immobile inclusions, indicative of aggregated proteins, occur (van Ham et al., Reference van Ham, Thijssen, Breitling, Hofstra, Plasterk and Nollen2008). These results were confirmed recently by FLIM (Table 1), in which reduction of the fluorescence lifetime of the YFP tag, indicative of fibril formation by α-synuclein, is only observed after day 10 (Laine et al., Reference Laine, Sinnige, Ma, Haack, Poudel, Gaida, Curry, Perni, Nollen, Dobson, Vendruscolo, Kaminski Schierle and Kaminski2019). However, motility defects are observed from day 4 onwards (van Ham et al., Reference van Ham, Thijssen, Breitling, Hofstra, Plasterk and Nollen2008), suggesting that inclusion formation generates toxicity prior to the formation of detectable amyloid-like aggregates.
C. elegans has a particular advantage for PD research in that it contains eight dopaminergic neurons that can easily be tracked. Models overexpressing wild-type and mutant α-synuclein in dopaminergic neurons have been generated in several independent studies (Lakso et al., Reference Lakso, Vartiainen, Moilanen, Sirviö, Thomas, Nass, Blakely and Wong2003; Cao et al., Reference Cao, Gelwix, Caldwell and Caldwell2005; Kuwahara et al., Reference Kuwahara, Koyama, Gengyo-Ando, Masuda, Kowa, Tsunoda, Mitani and Iwatsubo2006). Although inclusions have only been found in a small fraction of neurons (Lakso et al., Reference Lakso, Vartiainen, Moilanen, Sirviö, Thomas, Nass, Blakely and Wong2003; Kuwahara et al., Reference Kuwahara, Koyama, Gengyo-Ando, Masuda, Kowa, Tsunoda, Mitani and Iwatsubo2006), or not at all (Cao et al., Reference Cao, Gelwix, Caldwell and Caldwell2005), all studies report a loss of neuronal integrity. Expression of A53T and A30P, but not wild-type, α-synuclein was found to be associated with a loss of dendrites, in agreement with behavioural defects in food sensing, which depends on dopaminergic neurons (Kuwahara et al., Reference Kuwahara, Koyama, Gengyo-Ando, Masuda, Kowa, Tsunoda, Mitani and Iwatsubo2006). This phenotype has been observed in 3-day old animals (Kuwahara et al., Reference Kuwahara, Koyama, Gengyo-Ando, Masuda, Kowa, Tsunoda, Mitani and Iwatsubo2006), and in 4-day old animals in another study (Lakso et al., Reference Lakso, Vartiainen, Moilanen, Sirviö, Thomas, Nass, Blakely and Wong2003), suggesting that neurotoxicity occurs relatively early in the lifespan of the animals. Furthermore, these studies suggest that α-synuclein neurotoxicity is not correlated with inclusion formation, but may be caused by the presence of smaller aggregates or soluble oligomeric species of α-synuclein.
Fly models
Expression of wild-type, A53T and A30P α-synuclein in the nervous system of Drosophila was found to lead to the formation of age-related Lewy body-like inclusions, correlated with the loss of dopaminergic neurons and impaired climbing function (Feany and Bender, Reference Feany and Bender2000). In another model expressing the same α-synuclein variants specifically in the dopaminergic neurons, neuronal loss was also observed which could be restored by overexpression of the molecular chaperone Hsp70 (Auluck et al., Reference Auluck, Chan, Trojanowski, Lee and Bonini2002). Some controversy remains, however, about the extent of neuronal loss in fly models of PD, and other groups who used different methods have reported that the neurons might have been dysfunctional instead of lost (Pesah et al., Reference Pesah, Burgess, Middlebrooks, Ronningen, Prosser, Tirunagaru, Zysk and Mardon2005; Botella, Bayersdorfer and Schneuwly, Reference Botella, Bayersdorfer and Schneuwly2008). The inclusions in the original model were examined by EM to reveal mostly round aggregates consisting of a rather homogeneous core, surrounded by radiating filaments (Feany and Bender, Reference Feany and Bender2000). The model also displays diffuse aggregates and neuritic threads. Perhaps surprisingly, no differences between the phenotype of the wild-type and mutant α-synuclein lines have been found in this study (Feany and Bender, Reference Feany and Bender2000). The severity of the phenotype may correlate better with the α-synuclein expression levels, given that a more recent model achieving higher expression levels in the fly brain using the Q binary expression system shows exacerbated locomotory defects and a shortened lifespan, accompanied by α-synuclein inclusions that are mostly ubiquitin-positive, and vacuolar neurodegeneration (Ordonez et al., Reference Ordonez, Lee and Feany2018).
The relationship between α-synuclein aggregation and neurotoxicity has been further addressed by models expressing truncated or modified forms of the protein. A construct lacking part of the non-amyloid component (NAC) domain, which is essential for α-synuclein aggregation (Fig. 2c), does not induce either inclusion formation or degeneration of dopaminergic neurons, whereas a more aggregation-prone C-terminal truncation mutant shows increased deposition of proteins resistant to proteinase K (PK), as well as higher toxicity, supporting a direct correlation between α-synuclein aggregation and toxicity (Periquet et al., Reference Periquet, Fulga, Myllykangas, Schlossmacher and Feany2007). Notably, analysis by western blot has indicated that the levels of soluble oligomers are decreased in the NAC deletion flies, and increased in the C-terminal mutant line, leaving the question open as to whether the PK-resistant species or the soluble oligomers are responsible for toxicity (Periquet et al., Reference Periquet, Fulga, Myllykangas, Schlossmacher and Feany2007).
Other studies have shown that the presence of large inclusions does not correlate with toxicity, and may even be protective. When phosphorylation of S129 is blocked by the mutation of serine to alanine, more inclusions are formed, correlating with more PK-resistant protein, but a reduced loss of dopaminergic neurons (Chen and Feany, Reference Chen and Feany2005). How this observation relates to PD in humans is unclear, as S129-phosphorylated α-synuclein is found in Lewy bodies (Fujiwara et al., Reference Fujiwara, Hasegawa, Dohmae, Kawashima, Masliah, Goldberg, Shen, Takio and Iwatsubo2002). However, a subsequent study has shown that S129 phosphorylation increases oligomer formation, explaining the exacerbated phenotype in the fly model, whereas phosphorylation at Y215 is protective, as blocking this modification results in increased oligomer levels and toxicity (Chen et al., Reference Chen, Periquet, Wang, Negro, McLean, Hyman and Feany2009).
The role of α-synuclein oligomers has been further addressed by designing proline mutants that prevent β-strand formation in α-synuclein (Karpinar et al., Reference Karpinar2009). These mutations have been found to inhibit fibril formation in vitro, yet increase the generation of oligomeric species. When expressed in C. elegans vulval muscles, wild-type α-synuclein-YFP forms elongated structures, whereas a triple proline mutant shows a diffuse localisation consistent with the absence of large aggregates (Karpinar et al., Reference Karpinar2009). However, the triple proline mutant shows an increased toxicity in both worm and fly models when expressed in dopaminergic neurons, supporting a role for α-synuclein oligomers in neurotoxicity (Karpinar et al., Reference Karpinar2009).
Rodent and primate models
As described above in the context of AD models, it has been challenging to develop a rodent model that recapitulates the main characteristic features of PD, including Lewy body pathology, specific loss of dopaminergic neurons in the substantia nigra, and loss of motor function. A complication arises from the fact that mice and rats express their endogenous forms of α-synuclein, which differ from the human form at seven residues, one of the differences being the A53T substitution, which in humans is associated with familial PD. Here, we discuss rodent models based on the transgenic expression of human α-synuclein, focusing on its misfolding and aggregation and the associated phenotypes (Table 5). For a more complete description, see Fernagut and Chesselet (Reference Fernagut and Chesselet2004); Dehay and Fernagut (Reference Dehay and Fernagut2016) and Visanji et al. (Reference Visanji, Brotchie, Kalia, Koprich, Tandon, Watts and Lang2016), and for a discussion on models designed to study the spreading of α-synuclein pathology from cell-to-cell in a prion-like fashion, which is thought to play a role in synucleinopathies, we refer to Dehay et al. (Reference Dehay, Vila, Bezard, Brundin and Kordower2016).
The first-transgenic mouse model expressing human wild-type α-synuclein, which used the PDGF-β promoter, showed accumulation of the protein in several brain regions, including the neocortex, hippocampus and substantia nigra (Masliah et al., Reference Masliah, Rockenstein, Veinbergs, Mallory, Hashimoto, Takeda, Sagara, Sisk and Mucke2000). However, these inclusions did not resemble Lewy bodies, appearing in the nucleus as well as the cytoplasm, being associated with the ER, having a less well-defined shape, and consisting of granular rather than fibrillar material. Use of the Thy1 promoter led to a different spatial expression pattern of wild-type α-synuclein (Rockenstein et al., Reference Rockenstein, Mallory, Hashimoto, Song, Shults, Lang and Masliah2002), and extensive characterisation of this model, termed Line 61, has revealed PK-resistant inclusions, phosphorylation of S129, reduced dopamine levels, and progressive PD-like symptoms including olfactory deficits and locomotory impairments, but no substantial neuronal loss (Chesselet et al., Reference Chesselet, Richter, Zhu, Magen, Watson and Subramaniam2012). A model expressing A53T α-synuclein under control of the strong PrP promoter showed insoluble, fibrillar protein and inclusions that stain for ThS, in contrast to the wild-type protein (Giasson et al., Reference Giasson, Duda, Quinn, Zhang, Trojanowski and Lee2002). At 8 months of age, the A53T mice start to develop motor defects, whereas wild-type α-synuclein-transgenic mice are unaffected. However, the A53T phenotype is not associated with a loss of dopaminergic neurons in the substantia nigra in this model (Giasson et al., Reference Giasson, Duda, Quinn, Zhang, Trojanowski and Lee2002).
Conversely, other transgenic models led to degeneration of dopaminergic neurons in the substantia nigra but without the presence of observable inclusions, as exemplified by models expressing a combined A30P + A53T mutant (Richfield et al., Reference Richfield, Thiruchelvam, Cory-Slechta, Wuertzer, Gainetdinov, Caron, Di Monte and Federoff2002; Thiruchelvam et al., Reference Thiruchelvam, Powers, Cory-Slechta and Richfield2004), and a C-terminally truncated construct (Wakamatsu et al., Reference Wakamatsu, Ishii, Iwata, Sakagami, Ukai, Ono, Kanbe, Muramatsu, Kobayashi, Iwatsubo and Yoshimoto2008), respectively. In a model expressing wild-type human α-synuclein from a bacterial artificial chromosome (BAC) construct that includes the native human regulatory sequences, defects in dopaminergic neurotransmission are accompanied by the presence of high-molecular weight soluble species, although PK-resistant aggregates have not been identified, suggesting that toxicity may arise from intermediate species along the aggregation pathway (Janezic et al., Reference Janezic, Threlfell, Dodson, Dowie, Taylor, Potgieter, Parkkinen, Senior, Anwar, Ryan, Deltheil, Kosillo, Cioroch, Wagner, Ansorge, Bannerman, Bolam, Magill, Cragg and Wade-Martins2013). A mouse model with inducible A53T α-synuclein expression similarly shows high-molecular weight species on western blots in the absence of large inclusions, which correlate with perturbations in the Golgi apparatus in midbrain dopaminergic neurons, as well as changes in lysosomal and autophagic markers (Lin et al., Reference Lin, Parisiadou, Sgobio, Liu, Yu, Sun, Shim, Gu, Luo, Long, Ding, Mateo, Sullivan, Wu, Goldstein, Lovinger and Cai2012).
Recently, a mouse model was created following the hypothesis that native α-synuclein can occur as folded tetramers, such that a shift to the monomeric conformation puts the protein at risk of aggregation (Bartels et al., Reference Bartels, Choi and Selkoe2011; Dettmer et al., Reference Dettmer, Newman, Soldner, Luth, Kim, Von Saucken, Sanderson, Jaenisch, Bartels and Selkoe2015). Overexpression of a triple mutant thought to disrupt the tetramers, referred to as 3K because of the three E-to-K substitutions, gives rise to a strong motor phenotype, selective degeneration of dopaminergic neurons in the substantia nigra, and abnormal accumulation of α-synuclein co-localising with vesicles (Nuber et al., Reference Nuber, Rajsombath, Minakaki, Winkler, Müller, Ericsson, Caldarone, Dettmer and Selkoe2018). When compared at similar expression levels, the phenotype of the 3K mutant mice is stronger than that of a model expressing a BAC construct with the single E46K mutant that is found in some cases of familial PD. In addition, 3K is present in C-terminally truncated and S129 phosphorylated forms, and in older mice fibrillar material is present in the α-synuclein/vesicle assemblies (Nuber et al., Reference Nuber, Rajsombath, Minakaki, Winkler, Müller, Ericsson, Caldarone, Dettmer and Selkoe2018), reminiscent of recent investigations showing that Lewy bodies contain vesicular structures (Shahmoradian et al., Reference Shahmoradian, Lewis, Genoud, Hench, Moors, Navarro, Castaño-Díez, Schweighauser, Graff-Meyer, Goldie, Sütterlin, Huisman, Ingrassia, de Gier, Rozemuller, Wang, Paepe, De Erny, Staempfli, Hoernschemeyer, Großerüschkamp, Niedieker, El-Mashtoly, Quadri, Van IJcken, Bonifati, Gerwert, Bohrmann, Frank, Britschgi, Stahlberg, Van de Berg and Lauer2019; Mahul-Mellier et al., Reference Mahul-Mellier, Burtscher, Maharjan, Weerens, Croisier, Kuttler, Leleu, Knott and Lashuel2020).
Applying the BAC approach to create transgenic rats has led to a model that also recapitulates these and other features of human PD (Nuber et al., Reference Nuber, Harmuth, Kohl, Adame, Trejo, Schönig, Zimmermann, Bauer, Casadei, Giel, Calaminus, Pichler, Jensen, Müller, Amato, Kornhuber, Teismann, Yamakado, Takahashi, Winkler, Masliah and Riess2013). Full-length as well as C-terminally truncated insoluble α-synuclein accumulate as the rats age, following a similar development to that observed in the Braak stages of PD (Braak et al., Reference Braak, Del Tredici, Rüb, de Vos, Jansen Steur and Braak2003). PK-resistant inclusions are found in several brain regions including the substantia nigra, and the presence of aggregated protein has been further confirmed by dot blots using the FILA-1 antibody which recognises α-synuclein oligomers and fibrils but not monomers (Nuber et al., Reference Nuber, Harmuth, Kohl, Adame, Trejo, Schönig, Zimmermann, Bauer, Casadei, Giel, Calaminus, Pichler, Jensen, Müller, Amato, Kornhuber, Teismann, Yamakado, Takahashi, Winkler, Masliah and Riess2013). Electron micrographs have shown inclusions containing lysosomes and lipid droplets, as well as accumulated vesicles at synaptic termini. In addition to the accumulation of α-synuclein, the phenotype of this rat model also recapitulates certain aspects of the course of PD in humans, with early changes in avoidance and novelty-seeking behaviour and defects in the sense of smell, followed by later motor symptoms, which correlate with the loss of dopaminergic neurons (Nuber et al., Reference Nuber, Harmuth, Kohl, Adame, Trejo, Schönig, Zimmermann, Bauer, Casadei, Giel, Calaminus, Pichler, Jensen, Müller, Amato, Kornhuber, Teismann, Yamakado, Takahashi, Winkler, Masliah and Riess2013).
The injection of α-synuclein constructs in lentiviral or recombinant adeno-associated viral (rAAV) vectors has been used as an alternative approach to generate high local concentrations of the protein in the substantia nigra and to induce rapid neurodegeneration specifically in this region. The majority of viral vector rat and mouse models displays α-synuclein accumulation in the cytoplasm as well as in dystrophic neurites, yet does not feature Lewy body-like pathology (Kirik et al., Reference Kirik, Rosenblad, Burger, Lundberg, Johansen, Muzyczka, Mandel and Björklund2002; Lo Bianco et al., Reference Lo Bianco, Ridet, Schneider, Deglon and Aebischer2002; Chung et al., Reference Chung, Koprich, Siddiqi and Isacson2009; Dehay and Fernagut, Reference Dehay and Fernagut2016). However, PK-resistant α-synuclein and small ThS-positive inclusions have been observed in a rat rAAV model (da Silveira et al., Reference da Silveira, Schneider, Cifuentes-Diaz, Sage, Abbas-Terki, Iwatsubo, Unser and Aebischer2009). α-Synuclein accumulates close to the nucleus, the ER and Golgi in this model, and some fibrillar structures were observed in electron micrographs to co-localise with lysosomes and other vesicles. Interestingly, the number of ThS-positive inclusions was significantly increased when using the S129A α-synuclein mutation, which cannot be phosphorylated (da Silveira et al., Reference da Silveira, Schneider, Cifuentes-Diaz, Sage, Abbas-Terki, Iwatsubo, Unser and Aebischer2009), an observation analogous to the results obtained in the Drosophila model (Chen and Feany, Reference Chen and Feany2005). On the other hand, the phosphomimetic S129D mutation displays fewer, but larger ThS-positive aggregates, whereas no difference in toxicity was found between the different phosphorylation states (da Silveira et al., Reference da Silveira, Schneider, Cifuentes-Diaz, Sage, Abbas-Terki, Iwatsubo, Unser and Aebischer2009). However, another study using rat rAAV models reports that the non-phosphorylatable S129A mutant causes more dopaminergic neurodegeneration, whereas S129D forms more inclusions, although the latter have not been examined ultrastructurally (Gorbatyuk et al., Reference Gorbatyuk, Li, Sullivan, Chen, Kondrikova, Manfredsson, Mandel and Muzyczka2008). The precise role of S129 phosphorylation in α-synuclein aggregation and toxicity thus remains to be determined (Sato et al., Reference Sato, Kato and Arawaka2013).
Viral vector models have provided a unique means to assess the effect of varying α-synuclein concentrations on its aggregation and the time course of neurodegeneration following injection. The appearance of ThS-positive α-synuclein A53T inclusions is dose-dependent and time-dependent in rats, and correlates with dopaminergic dysfunction (Van der Perren et al., Reference Van der Perren, Toelen, Casteels, Macchi, Van Rompuy, Sarre, Casadei, Nuber, Himmelreich, Osorio Garcia, Michotte, D'Hooge, Bormans, Van Laere, Gijsbers, Van den Haute, Debyser and Baekelandt2015). Western blots have confirmed that insoluble and phosphorylated α-synuclein accumulates over time, and that high-molecular weight oligomeric α-synuclein species are apparent at later time points after injection (Van der Perren et al., Reference Van der Perren, Toelen, Casteels, Macchi, Van Rompuy, Sarre, Casadei, Nuber, Himmelreich, Osorio Garcia, Michotte, D'Hooge, Bormans, Van Laere, Gijsbers, Van den Haute, Debyser and Baekelandt2015). Using the same type of viral vector, a mouse model has also been established that displays Lewy body-like pathology and degeneration of dopaminergic neurons, phosphorylation of S129 and impairments in motor behaviour (Oliveras-Salvá et al., Reference Oliveras-Salvá, Van Der Perren, Casadei, Stroobants, Nuber, D'Hooge, Van Den Haute and Baekelandt2013). Both wild-type and A53T α-synuclein lead to similar levels of protein accumulation and loss of dopaminergic neurons using this approach. Another type of optimised rAAV vector has also been employed successfully in mice to express A53T α-synuclein locally leading to a robust phenotype, including the loss of dopaminergic neurons and reduced dopamine levels, accompanied by the formation of PK-resistant α-synuclein inclusions in cell bodies and dystrophic neurites, resembling PD pathology (Ip et al., Reference Ip, Klaus, Karikari, Visanji, Brotchie, Lang, Volkmann and Koprich2017).
The success with rAAV vectors in rodents has led to the development of non-human primate models using the same technology, resulting in a marmoset model that features α-synuclein accumulation in the cytoplasm and neurites, as well as partial dopaminergic neurodegeneration and motor symptoms (Kirik et al., Reference Kirik, Annett, Burger, Muzyczka, Mandel and Bjorklund2003). In a subsequent study, the monkeys were followed over a longer period of up to a year, leading to the discovery of ubiquitin-positive inclusions, which also occur in human PD (Eslamboli et al., Reference Eslamboli, Romero-Ramos, Burger, Bjorklund, Muzyczka, Mandel, Baker, Ridley and Kirik2007). Furthermore, animals injected with the A53T α-synuclein construct show stronger neurodegeneration compared to the wild-type protein and a worsened phenotype at later time points, potentially making this a useful model to test therapeutic strategies.
Identification of biological pathways using in vivo models
Several current disease models are successful in recapitulating key aspects of protein misfolding and aggregation, and exhibit toxic phenotypes, making them useful to interrogate disease mechanisms by identifying the perturbed cellular pathways. Excellent tools are available for commonly used model organisms to probe these questions at the system level by means of genomic, transcriptomic and proteomic studies.
Simple model organisms, including yeast, worms and flies, are amenable to large screens to identify genetic modifiers of protein aggregation and the associated toxicity. In addition, transcriptomics and proteomics can be applied to any organism of which the RNA or protein content can be extracted, to inform on the dysregulation of cellular pathways upon expression of the disease-related gene. Improvements in sequencing technologies have recently increased dramatically the scope of transcriptomics studies, which were previously limited to microarray analysis on a predefined set of genes, whereas genome-wide expression levels can now be determined routinely with high sensitivity and few artefacts using next-generation sequencing methods such as RNAseq (Wang et al., Reference Wang, Gerstein and Snyder2009; Breschi et al., Reference Breschi, Gingeras and Guigó2017). Similarly, developments in mass spectrometry and methods for data analysis have greatly enhanced the possibilities of performing comparative and quantitative proteomics to identify changes in protein abundance as well as post-translational modifications (Bantscheff et al., Reference Bantscheff, Lemeer, Savitski and Kuster2012; Feng et al., Reference Feng, Cappelletti and Picotti2017; Schubert et al., Reference Schubert, Röst, Collins, Rosenberger and Aebersold2017). Mass spectrometers have not only improved in terms of sensitivity and resolution, but also the opportunities for quantitative analysis of animal and human samples have expanded significantly by chemical labelling approaches such as tandem mass tags (TMTs) and isobaric tags for absolute and relative quantification (iTRAQ). Alternatively, label-free approaches based on counting the number of peptide-to-spectrum matches for each protein have gained popularity. In addition, dedicated proteomic workflows exist that inform on protein–protein interactions and on protein conformations, although these methods are currently largely restricted to unicellular organisms and cultured cells (Minde et al., Reference Minde, Dunker and Lilley2017).
In this section, we highlight the key pathways and genes that have been identified using these various technologies in relation to protein misfolding in the organismal models of AD and PD (Tables 2–5).
Biological pathways in Alzheimer's disease models
Yeast models
Of the in vivo models described here, yeast is perhaps the most highly amenable to systematic analysis at the levels of the genome, transcriptome and proteome. Extensive toolkits are available, and profiling of genetic interactions, mRNA expression levels, protein levels, metabolite levels, biological effects of small molecules and phenotypic responses can be performed on a large scale fairly readily (Khurana and Lindquist, Reference Khurana and Lindquist2010). Although yeast does have a nervous system, or the secretase activity required for APP processing, it has been used to identify conserved-genetic factors involved in Aβ toxicity (Treusch et al., Reference Treusch, Hamamichi, Goodman, Matlack, Chung, Baru, Shulman, Parrado, Bevis, Valastyan, Han, Lindhagen-persson, Reiman, Evans, Bennett, Olofsson, Dejager, Tanzi, Caldwell, Caldwell and Lindquist2011). The most potent suppressors of such toxicity were found to be genes involved in endocytosis, including the yeast homologue of PICALM, which is a known risk factor for AD as identified by genome-wide association studies (GWAS). A number of these hits have been confirmed in a C. elegans model expressing Aβ42 in glutamatergic neurons (Treusch et al., Reference Treusch, Hamamichi, Goodman, Matlack, Chung, Baru, Shulman, Parrado, Bevis, Valastyan, Han, Lindhagen-persson, Reiman, Evans, Bennett, Olofsson, Dejager, Tanzi, Caldwell, Caldwell and Lindquist2011). Microarray analysis revealed the induction of ER stress and downregulation of mitochondrial processes upon expression of Aβ42 targeted to the secretory pathway (Chen et al., Reference Chen, Bisschops, Agarwal, Ji, Shanmugavel and Petranovic2017).
Worm models
The main power of C. elegans in genetic screens is the ease with which RNA interference (RNAi) can be performed, simply by feeding the worms with bacteria expressing the relevant double-stranded RNA (dsRNA). Libraries targeting 86% of all the annotated C. elegans genes are available and can be routinely screened if an appropriate read-out is available (Sin et al., Reference Sin, Michels and Nollen2014). Performing RNAi by feeding is very efficient for certain tissues, such as intestinal and muscle cells, but works less well in neurons. With respect to Aβ toxicity, C. elegans has been used for an RNAi screen specifically targeted at a conserved set of molecular chaperones (Brehme et al., Reference Brehme, Voisine, Rolland, Wachi, Soper, Zhu, Orton, Villella, Garza, Vidal, Ge and Morimoto2014). Out of 219 molecular chaperone genes tested, knockdown of 18 led to increased paralysis rates in animals expressing Aβ in the muscle cells. The majority of these genes are also protective in worms expressing polyglutamine, the aggregation of which is linked to Huntington's disease and a variety of other conditions, indicating that their effects may be related to a general improvement in protein homoeostasis capacity (Brehme et al., Reference Brehme, Voisine, Rolland, Wachi, Soper, Zhu, Orton, Villella, Garza, Vidal, Ge and Morimoto2014). Another RNAi screen in the C. elegans model expressing Aβ in the muscle cells was targeted at genes with known human homologues, leading to the identification of various hits, including subunits of a vacuolar ATPase and of the ribosome, and a component of the TRiC/CCT chaperone (Khabirova et al., Reference Khabirova, Moloney, Marciniak, Williams, Lomas, Oliver, Favrin, Sattelle and Crowther2014). Mitochondrial function was furthermore shown to be impaired in a model expressing Aβ42 in the neurons (Fong et al., Reference Fong, Teo, Ng, Chen, Lakshmanan, Tsoi, Moore, Inoue, Halliwell and Gruber2016), and metabolomics and transcriptomics subsequently demonstrated that early changes in tricarboxylic acid cycle metabolism were underlying this phenotype (Teo et al., Reference Teo, Ravi, Barardo, Kim, Fong, Cazenave-Gassiot, Tan, Ching, Kovalik, Wenk, Gunawan, Moore, Halliwell, Tolwinski and Gruber2019).
The link between Aβ accumulation and the action of molecular chaperones was further supported by mass spectrometry, in a study where two Hsp70 proteins and three small heat shock proteins (sHSPs) were found to co-localise with Aβ deposits in the body wall muscle cells of C. elegans (Fonte et al., Reference Fonte, Kapulkin, Taft, Fluet, Friedman and Link2002). In a study on an inducible C. elegans model expressing Aβ in body wall muscle cells, gene expression changes assessed by RNA microarrays also include sHSPs and Hsp70, as well as mitochondrial unfolded protein response (UPR) genes and proteasomal factors. An aggregation-prone GFP variant was employed as a control in this study, enabling the identification of the genes that are differentially expressed specifically as a consequence of Aβ-mediated toxicity, rather than resulting from protein aggregation in general (Hassan et al., Reference Hassan, Dostal, Huemann, Yerg and Link2015). The changes in gene expression resemble those induced by a pore-forming toxin, Cry5B, suggesting that Aβ may exert its toxic effects in this model by affecting membrane integrity.
In a genome-wide screen in C. elegans expressing mutant tau in neurons, molecular chaperones have been identified along with proteases, kinases and phosphatases, and a number of other enzymes, signalling proteins and DNA/RNA regulating factors (Kraemer et al., Reference Kraemer, Burgess, Chen, Thomas and Schellenberg2006). The identification of kinases and phosphatases is consistent with the known importance of tau phosphorylation in AD. However, RNAi knockdown of the C. elegans homologues of GSK3-β and MARK worsens the phenotype of the tau model, suggesting that the action of these kinases is beneficial, in contrast to their role in tau pathology in humans (Hanger et al., Reference Hanger, Anderton and Noble2009).
Fly models
Drosophila-genetic screens are often based on screening for suppressors or enhancers of the rough eye phenotype caused by the expression of proteins that give rise to toxicity. Fly genetics have provided excellent tools for genome-wide studies, e.g. using randomly inserted transposable elements containing a upstream activation sequence that drives gene expression upon activation by the GAL4 protein, which can be expressed in a tissue-specific manner. Depending on the location and direction of insertion of the transposable element in the context of a gene, expression can either be enhanced or disrupted. Using this system, a screen has been performed on flies expressing Aβ42 in the eye, leading to the identification of 23 modifier genes of which several have been confirmed by mutagenesis (Cao et al., Reference Cao, Song, Gangi, Kelkar, Antani, Garza and Konsolaki2008). Consistent with the role of these pathways in human disease, genes operating in the secretory pathway and involved in cholesterol metabolism were found, as well as genes regulating chromatin structure and function (Cao et al., Reference Cao, Song, Gangi, Kelkar, Antani, Garza and Konsolaki2008). In a complementary approach using a loss-of-function library, the gene Toll has been identified, which bears homology to the mammalian interleukin-1 receptor. Deletion of this gene or mutations in downstream-signalling components ameliorate the rough eye phenotype and shorten the lifespan of the Aβ42 flies, suggesting an important role of immune signalling in Aβ42-mediated toxicity (Tan et al., Reference Tan, Schedl, Song, Garza and Konsolaki2008).
A microarray transcriptomics study on flies expressing Aβ42 throughout the nervous system revealed changes in pathways related to oxidative stress and the immune response (Favrin et al., Reference Favrin, Bean, Bilsland, Boyer, Fischer, Russell, Crowther, Baylis, Oliver and Giannakou2013). Although these pathways are also altered during the ageing of control flies, several genes are affected specifically by the expression of Aβ42, such as the superoxide dismutase gene Sod3 which is upregulated ~8-fold compared to controls. Further changes in the Aβ42 flies include the upregulation of a gene involved in ER-to-Golgi transport, and the downregulation of a putative ER molecular chaperone, consistent with the involvement of the secretory pathway in Aβ42 toxicity (Favrin et al., Reference Favrin, Bean, Bilsland, Boyer, Fischer, Russell, Crowther, Baylis, Oliver and Giannakou2013).
Drosophila tau models have yielded a set of pathways related to toxicity that are different from those in the Aβ models. Genetic screens using libraries of transposable element insertions led to the identification of phosphatases and kinases as modifiers of human tau toxicity in the fly eye (Shulman and Feany, Reference Shulman and Feany2003; Ambegaokar and Jackson, Reference Ambegaokar and Jackson2011), as well as diverse pathways including autophagy and lysosomal clearance, RNA processing, chromatin regulation, molecular chaperones and the cytoskeleton (Ambegaokar and Jackson, Reference Ambegaokar and Jackson2011). An RNAi screen on a tau R406W model also highlighted the role of the cytoskeleton, in particular dynein/dynactin-mediated retrograde axonal transport, in addition to the activity of kinases and phosphatases, and protein metabolism (Butzlaff et al., Reference Butzlaff, Hannan, Karsten, Lenz, Ng, Voßfeldt, Prüßing, Pflanz, Schulz, Rasse and Voigt2015). Although the role of kinases and phosphatases seems obvious, given the relationship between the phosphorylation state of tau and its propensity to aggregate and cause toxicity, the genetic interactions were shown to be rather complex (Ambegaokar and Jackson, Reference Ambegaokar and Jackson2011). The phosphorylation state of tau, as probed with the AT8 antibody, does not correlate with either enhancers or suppressors of aggregation. Furthermore, the reduced expression of the GSK3-β homologue is a potent suppressor not only of tau toxicity but also of that induced by Aβ42 and TDP-43, suggesting that the network of kinases and phosphatases may play a role in common mechanisms underlying neurodegeneration (Ambegaokar and Jackson, Reference Ambegaokar and Jackson2011).
The Drosophila model based on tau V337M has been used to validate hits from GWAS performed on human AD patients. In one study, the relatively small number of participants did not produce statistically significant results, yet 6 out of 19 Drosophila homologues of candidate genes that were tested were shown to be modifiers of tau toxicity in the fly eye, including the glucose transporter SLC2A14 (Shulman et al., Reference Shulman, Chipendo, Chibnik, Aubin, Tran, Keenan, Kramer, Schneider, Bennett, Feany and De Jager2011). In a follow-up study of a larger number of GWAS-identified loci, 9 out of 84 genes were confirmed to be modifiers of toxicity including two genes involved in cell adhesion, CD2AP and FERMT2 (Shulman et al., Reference Shulman, Imboywa, Giagtzoglou, Powers, Hu, Devenport, Chipendo, Chibnik, Diamond, Perrimon, Brown, De Jager and Feany2014). Thus, this screening strategy appears to have a high success rate compared to that of unbiased screens, and can validate GWAS results below the threshold for statistical significance. It will be interesting to observe whether or not Drosophila models based on Aβ42 can confirm the roles of additional associated genes. A limitation is, however, presented by human genes which do not have Drosophila homologues, such as those involved in lipid metabolism (APOE and CLU) and immunity (CR1, CD33 and MS4A) that are strongly associated with the risk of late-onset AD (Shulman et al., Reference Shulman, Imboywa, Giagtzoglou, Powers, Hu, Devenport, Chipendo, Chibnik, Diamond, Perrimon, Brown, De Jager and Feany2014).
Rodent models
Genetic screening for modifiers of protein misfolding is not commonly undertaken in rodents, but analysis of gene expression levels and of protein abundance have been applied extensively to mouse models of AD to shed light on the cellular pathways that are disturbed in response to protein aggregation. Especially in recent years, methods of analysing gene expression using novel sequencing technologies, as well as methods in mass spectrometry to detect protein levels and modifications, have greatly expanded these possibilities.
A meta-analysis of the transcriptomes of aged and diseased humans versus mice has revealed that changes during ageing are relatively well-conserved between the two species, for example consistently showing upregulation of inflammatory factors (Hargis and Blalock, Reference Hargis and Blalock2017). On the other hand, the gene expression profiles of mouse models of neurodegenerative diseases such as AD compared to healthy littermates display limited overlap with those of human patients (Burns et al., Reference Burns, Li, Mehta, Awad and Morgan2015; Hargis and Blalock, Reference Hargis and Blalock2017), showing, for example, less downregulation of genes associated with mitochondrial function, protein degradation, RNA processing and DNA repair (Burns et al., Reference Burns, Li, Mehta, Awad and Morgan2015). Although the differences in gene expression between the animal models and human patients represent a major obstacle for the screening of drug candidates in preclinical trials, the models do allow perturbations in response to the misfolding of transgenically expressed proteins to be deciphered. Complementary insights into the dysregulation of cellular pathways can be obtained from the quantification of protein abundance, which does not always correlate directly with gene expression levels (Liu et al., Reference Liu, Beyer and Aebersold2016), especially under conditions where protein quality control and turnover are perturbed. In the case of AD, transcriptomics and proteomics studies have together pointed towards a consistent set of dysregulated pathways in various rodent models, including the immune response, mitochondrial function and synaptic transmission.
The activation of the immune response is generally thought to be a direct reaction to the deposition of Aβ plaques. In a comparative microarray analysis of brain tissue from Tg2576 and APPPS1 mice, the strongest change in gene expression was found to be a significant upregulation of the inflammatory factor progranulin, which was confirmed to be abundant in microglia and neurons in the vicinity of dense Aβ plaques (Pereson et al., Reference Pereson, Wils, Kleinberger, McGowan, Vandewoestyne, Van Broeck, Joris, Cuijt, Deforce, Hutton, Van Broeckhoven and Kumar-Singh2009). BRI-Aβ42 mice similarly display progranulin immune-reactivity surrounding Aβ plaques, suggesting a direct effect of extracellular plaque formation in triggering this immune response (Pereson et al., Reference Pereson, Wils, Kleinberger, McGowan, Vandewoestyne, Van Broeck, Joris, Cuijt, Deforce, Hutton, Van Broeckhoven and Kumar-Singh2009).
Another study has attempted to separate events resulting directly from Aβ plaque formation from those caused by other sources of toxicity by comparing the gene expression pattern of two models, the 5xFAD model which has an aggressive phenotype with early onset Aβ plaque formation, and a model expressing an N-terminal Aβ truncated form (Aβ4-42) that displays neuronal loss in the absence of Aβ plaques (Bouter et al., Reference Bouter, Kacprowski, Weissmann, Dietrich, Borgers, Brauß, Sperling, Wirths, Albrecht, Jensen, Kuss and Bayer2014). Despite the different pathologies, both models suffer from age-related memory and learning defects of similar magnitudes. In that study, deep sequencing revealed that more than half of the differentially expressed genes in aged mice expressing Aβ4-42 with respect to controls are similarly affected in the 5xFAD model, and that these are associated with in a broad range of biological functions including cell differentiation, neurogenesis, histone modification and transcription. Conversely, the majority of genes differentially expressed in the 5xFAD model compared to control mice are related to the immune response. These genes are not altered in the Aβ4-42 mice, suggesting that these perturbations are a direct consequence of reactivity to Aβ plaques in the 5xFAD model. Astrocytes are, however, activated in both models, as indicated by the upregulation of Gfap, encoding the typical marker glial fibrillary acidic protein (Bouter et al., Reference Bouter, Kacprowski, Weissmann, Dietrich, Borgers, Brauß, Sperling, Wirths, Albrecht, Jensen, Kuss and Bayer2014).
Microarray analysis of the gene expression profile of the 5xFAD mouse model also showed changes in genes involved in the immune response, pointing towards activation of the complement system and of interferon-γ regulated targets, concomitant with the appearance of pathological features including intracellular Aβ42 accumulation, Aβ plaque deposition and cognitive symptoms (Landel et al., Reference Landel, Baranger, Virard, Loriod, Khrestchatisky, Rivera, Benech and Féron2014). A mass spectrometric study specifically focusing on the activation of microglia has shown that the levels of the cytokines IL1β and IL10 are elevated before widespread plaque deposition occurs (Boza-Serrano et al., Reference Boza-Serrano, Yang, Paulus and Deierborg2018). A combined proteomics and genomics study on the 5xFAD mouse has also revealed changes in the expression of genes related to immune function, prior to the onset of Aβ plaque deposition (Bundy et al., Reference Bundy, Vied, Badger and Nowakowski2019). Other perturbations found in this study include the downregulation of proteins involved in the regulation of calcium binding, which show a trend in the same direction for mRNA and protein levels. A total of 40% of the genes, however, were found to show an anticorrelation with their corresponding protein levels, underscoring the potential differences between gene expression levels and protein abundance, and the importance of complementing transcriptomics with proteomics.
Given the evidence for a role of mitochondrial dysregulation in AD, a detailed proteomic analysis of mitochondria isolated from 3xTg mice has been performed, leading to the identification of 23 proteins that are either up- or down-regulated compared to non-transgenic mice (Chou et al., Reference Chou, Shenoy, Thomas, Choudhary, LaFerla, Goodman and Breen2011). These mitochondrial proteins have a variety of functions, including roles in metabolic processes such as oxidative phosphorylation but also functioning in ion transport, apoptosis and mitochondrial protein synthesis. The changes in protein levels are detectable in mice at 6 months of age, prior to widespread Aβ plaque deposition and tau tangle formation, thus reflecting a perturbation in mitochondrial homoeostasis early in the disease process (Chou et al., Reference Chou, Shenoy, Thomas, Choudhary, LaFerla, Goodman and Breen2011). A similar approach on the APPPS1 mouse model has revealed changes in the levels of 60 mitochondrial proteins with roles in the electron transport chain, citric acid cycle, oxidative stress and apoptosis, some of which are dysregulated prior to the onset of Aβ plaque formation (Völgyi et al., Reference Völgyi, Háden, Kis, Gulyássy, Badics, Györffy, Simor, Szabó, Janáky, Drahos, Dobolyi, Penke, Juhász and Kékesi2017).
An integrated proteomics and transcriptomics study on another APP/PSEN1 model took the approach of first investigating changes at the proteome level in different brain regions, followed by validation of the corresponding genetic pathways at the mRNA level (Kempf et al., Reference Kempf, Metaxas, Ibanez-Vea, Darvesh, Finsen and Larsen2016). Using mass spectrometry of peptides enriched in phosphorylation and glycosylation, changes in protein levels related to cytoskeleton-based synaptic plasticity were found, and the mRNA levels of upstream regulators such as glutamatergic receptors and neurotrophic factors were shown to be affected accordingly. Using quantitative mass spectrometry on APP, APP/PSEN1 and Bri-Aβ42 mice, proteins associated with late-onset AD were found to be altered, most notably ApoE, which was observed to be upregulated in all three models (Savas et al., Reference Savas, Wang, DeNardo, Martinez-Bartolome, McClatchy, Hark, Shanks, Cozzolino, Lavallée-Adam, Smukowski, Park, Kelly, Koo, Nakagawa, Masliah, Ghosh and Yates2017). Furthermore, proteins involved in excitatory synaptic transmission, in particular trafficking of the AMPA receptor, were found to be dysregulated, as well as mitochondrial proteins.
Several other reports have provided further evidence for the perturbation of synaptic plasticity at the proteome level. One study has specifically focused on the proteomic signature associated with memory defects, revealing an overlapping set of dysregulated membrane proteins involved in neuronal excitability and synaptic plasticity in the 5xFAD model of AD, as well as in aged control mice with memory impairment (Neuner et al., Reference Neuner, Wilmott, Hoffmann, Mozhui and Kaczorowski2017). Changes in the cytoskeleton as well as in synaptic function have been reported for the olfactory bulb of an APP/PSEN1 mouse model, occurring around the same age when Aβ plaques appear (Lachén-Montes et al., Reference Lachén-Montes, González-Morales, de Morentin, Pérez-Valderrama, Ausín, Zelaya, Serna, Aso, Ferrer, Fernández-Irigoyen and Santamaría2016). Levels of cytoskeletal proteins as well as chaperones have in addition been suggested to be modulated by the presence of Aβ oligomers, as discovered in the APP ΔE693 mouse strain that accumulates oligomers rather than plaques (Takano et al., Reference Takano, Yamashita, Nagano, Otani, Maekura, Kamada, Tsunoda, Tsutsumi, Tomiyama, Mori, Matsuura and Matsuyama2013). Finally, the McGill mutant APP rat model displays changes in proteins related to stress response and synaptic function at a young age before the onset of Aβ plaques, whereas at later age metabolism (e.g. mitochondrial function and amino acid metabolism) becomes affected (Do Carmo et al., Reference Do Carmo, Crynen, Paradis, Reed, Iulita, Ducatenzeiler, Crawford and Cuello2018).
Analysis of the gene expression patterns of tau-based-transgenic models has revealed important insights into the cellular responses towards tau aggregation. Transcriptional profiling of JNPL3 mice, which express tau P301L, revealed a down-regulation of anti-apoptotic factors, disturbances of microtubule-dependent intracellular trafficking and down-regulation of a number of immune factors (Ho et al., Reference Ho, Xiang, Mukherjee, Zhang, De Jesus, Mirjany, Yemul and Pasinetti2001). A study on another tau P301L model used more stringent selection criteria to identify just one upregulated gene, encoding glyoxalase I (Chen et al., Reference Chen, Wollmer, Hoerndli, Mu, Tsolaki and Papassotiropoulos2004). This enzyme functions in a pathway that prevents glycation and the formation of advanced glycated end products, which are known to occur in ageing and neurodegenerative disease. Glyoxalase I is strongly expressed in neurons with flame-shaped tau aggregates both in the mice and in AD patient brains, suggesting that it may be upregulated as a protective mechanism in response to protein aggregation (Chen et al., Reference Chen, Wollmer, Hoerndli, Mu, Tsolaki and Papassotiropoulos2004).
Changes in proteins functioning in metabolism, including mitochondrial proteins of the respiratory chain complex and detoxifiers of ROS, as well as those with a role in synaptic transmission have furthermore been detected in a proteomic analysis of the tau P301L mouse model (David et al., Reference David, Hauptmann, Scherping, Schuessel, Keil, Rizzu, Ravid, Dröse, Brandt, Müller, Eckert and Götz2005). The injection of Aβ42 fibrils into the amygdala of the tau P301L model has been found to result in further imbalances in components of the cytoskeleton, metabolism and synaptic transmission, as well as proteins functioning in stress response pathways and protein folding and degradation (David et al., Reference David, Ittner, Gehrig, Nergenau, Shepherd, Halliday and Götz2006). Many of these changes have also been found in SHSY-5Y cells expressing tau P301L upon injection with Aβ42 fibrils, suggesting conserved and rapid responses, whereas perturbations in the cytoskeleton and synaptic function appear exclusively in the mouse model, pointing towards longer-term or indirect consequences (David et al., Reference David, Ittner, Gehrig, Nergenau, Shepherd, Halliday and Götz2006). Proteomic analysis of postsynaptic density isolated from tau P301S mice has shown a depletion of GTPase-regulatory proteins, which could be related to the actin cytoskeletal defects and loss of dendritic spines that are observed in this model (Dejanovic et al., Reference Dejanovic, Huntley, De Mazière, Meilandt, Wu, Srinivasan, Jiang, Gandham, Friedman, Ngu, Foreman, Carano, Chih, Klumperman, Bakalarski, Hanson and Sheng2018). Consistent with many of these results, RNAseq of a more recent tau P301S mouse model has furthermore revealed perturbations in genes associated with presynaptic factors, glutamatergic signalling and protein scaffolding (Ke et al., Reference Ke2019).
An additive effect of Aβ and tau toxicity on mitochondrial dysfunction has been found in a triple mouse model, combining familial APP, PSEN2 and tau mutations (Rhein et al., Reference Rhein, Song, Wiesner, Ittner, Baysang, Meier, Ozmen, Bluethmann, Drose, Brandt, Savaskan, Czech, Gotz and Eckert2009). Mitochondrial proteins including Complex I and Complex IV subunits are downregulated at the protein level in the triple mouse model more strongly than in the parental APP/PSEN2 or tau strains. In support of these findings, the respiratory function of isolated mitochondria was found to be reduced more drastically and at earlier age in the triple model compared to the APP/PSEN2 strain or wild-type animals (Rhein et al., Reference Rhein, Song, Wiesner, Ittner, Baysang, Meier, Ozmen, Bluethmann, Drose, Brandt, Savaskan, Czech, Gotz and Eckert2009).
Proteomic approaches have moreover been used to assess the misfolding and aggregation of endogenous mouse proteins, which may be triggered by the pressure on the protein homoeostasis machinery caused by the aggregation of transgenic Aβ and tau. Analysis of the SDS-insoluble fraction from mice expressing APP with Swedish mutations K670N/M671L and PSEN1 Δexon9 has shown the presence of cytosolic proteins including glycolytic enzymes and members of the 14-3-3 family of chaperones (Xu et al., Reference Xu, Stevens, Moore, McClung and Borchelt2013). In an independent study on the same mouse model using quantitative proteomics, mitochondrial proteins have been identified in the sarkosyl-insoluble fraction of aged mice (Thygesen et al., Reference Thygesen, Metaxas, Larsen and Finsen2018). Finally, recent work on the inducible tau model rTg4510 has led to a set of SDS-insoluble proteins including kinases, other enzymes and chaperones that partially overlaps with that of the APP/PSEN1 model established using the same methods (Pace et al., Reference Pace, Xu, Fromholt, Howard, Crosby, Giasson, Lewis and Borchelt2018), suggesting a common mechanism that triggers the misfolding of otherwise soluble proteins.
Biological pathways in Parkinson's disease models
Yeast models
The genetic toolkit available for screens in yeast has been applied extensively to α-synuclein models, leading to the discovery of conserved cellular pathways involved in its toxicity. Early screens have made use of a genome-wide deletion library, showing an enrichment for genes involved in lipid metabolism and vesicle-mediated transport (Willingham et al., Reference Willingham, Outeiro, Devit, Lindquist, Muchowski, Willingham, Outeiro, Devit, Lindquist and Muchowski2003). In accordance with the latter, a genome-wide overexpression screen for suppressors and enhancers of α-synuclein toxicity identified the yeast homologue of human Rab1, a GTPase involved in ER-to-Golgi vesicle trafficking, as an important suppressor of toxicity. This protein is often found to co-localise with α-synuclein cytoplasmic inclusions, and its overexpression restores the trafficking defects associated with α-synuclein toxicity (Cooper et al., Reference Cooper, Gitler Cashikar, Cole, Haynes, Bhullar Liu, Xu, Strathearn, Liu, Cao, Caldwell, Caldwell, Marsischky, Kolodner, Labaer, Rochet, Bonini and Lindquist2006). Yeast homologues of Rab3a and Rab8a, which function in synaptic and post-Golgi vesicle transport, respectively, have also been found associated with the inclusions in subsequent work, and overexpression of any one of these three Rab proteins rescues toxicity in C. elegans expressing α-synuclein in dopaminergic neurons (Gitler et al., Reference Gitler, Bevis, Shorter, Strathearn, Hamamichi, Su, Caldwell, Caldwell, Rochet, McCaffery, Barlowe and Lindquist2008).
In addition to defects in ER-to-Golgi trafficking, mitochondrial dysfunction has been identified in a transcriptional screen of a yeast strain with high α-synuclein expression levels, together with an increase in ROS (Su et al., Reference Su, Auluck, Outeiro, Yeger-Iotem, Kritzer, Tardiff, Strathearn, Liu, Cao, Hamamichi, Hill, Caldwell, Bell, Fraenkel, Cooper, Caldwell, Mccaffery, Rochet and Lindquist2010). A library screen on this model has revealed a group of 1,2,3,4-tetrahydroquinolinones capable of reducing α-synuclein foci formation and toxicity, in addition to re-establishing ER-to-Golgi trafficking as well as mitochondrial function, suggesting that these processes are tightly linked. These compounds are furthermore active in a C. elegans model and in rat primary neuron cultures, providing evidence for a conserved mechanism (Su et al., Reference Su, Auluck, Outeiro, Yeger-Iotem, Kritzer, Tardiff, Strathearn, Liu, Cao, Hamamichi, Hill, Caldwell, Bell, Fraenkel, Cooper, Caldwell, Mccaffery, Rochet and Lindquist2010).
The role of vesicle trafficking has been confirmed once more in a study that made use of the high sensitivity to hydrogen peroxide of the yeast strain expressing α-synuclein (Liang et al., Reference Liang, Clark-Dixon, Wang, Flower, Williams-Hart, Zweig, Robinson, Tatchell and Witt2008). This study in addition reported ubiquitin-dependent protein degradation, protein biosynthesis and stress response genes as suppressors of toxicity. The YPP1 gene was discovered as a suppressor specifically for the α-synuclein A30P mutant, and the encoded protein was shown to target α-synuclein to vacuoles for degradation (Flower et al., Reference Flower, Clark-Dixon, Metoyer, Yang, Shi, Zhang and Witt2007). Findings of another overexpression screen using a large library of 5500 strains, covering 85% of the yeast genome, are largely in agreement with the pathways previously mentioned, in addition pointing to a role for protein phosphorylation in the toxicity of α-synuclein (Yeger-Lotem et al., Reference Yeger-Lotem, Riva, Su, Gitler, Cashikar, King, Auluck, Geddie, Valastyan, Karger, Lindquist and Fraenkel2009). In this screen, the yeast homologue of human PARK9 was also identified, which encodes a lysosomal P-type ATPase that is mutated in a familial form of PD (Gitler et al., Reference Gitler, Chesi, Geddie, Strathearn, Hamamichi, Hill, Caldwell, Caldwell, Cooper, Rochet and Lindquist2009). Overexpression of this gene suppresses α-synuclein toxicity not only in yeast, but also in a C. elegans model and in rat primary dopaminergic neurons. It also protects yeast cells against manganese-induced toxicity, a known risk factor for PD, providing a potential link between genetic and environmental factors in initiating the disease process (Gitler et al., Reference Gitler, Chesi, Geddie, Strathearn, Hamamichi, Hill, Caldwell, Caldwell, Cooper, Rochet and Lindquist2009). Two other hits from this screen, an E3 ubiquitin ligase and a factor related to nitrosative stress, have later been validated in cortical neurons differentiated from patient-derived induced pluripotent stem cells (iPSCs) (Chung et al., Reference Chung, Khurana, Auluck, Tardiff, Mazzuli, Soldner, Baru, Lou, Freyzon, Cho, Mungenast, Muffat, Mitalipova, Pluth, Jui, Schüle, Lippard, Tsai, Krainc, Buchwald, Jaenisch and Lindquist2013a).
Building on the results from genetic screens, a computational approach was designed with the aim of creating a genetic network underlying the disease phenotype, and of transposing it to the human homologues. Thus, a genetic map of α-synuclein toxicity has been created which spans the pathways identified previously, ranging from ER quality control and vesicle trafficking to calcium signalling and mitochondrial function, in which known genetic causes of familial PD appear in different parts of the network (Khurana et al., Reference Khurana2017). In an accompanying study that employs in situ peroxidase labelling coupled to mass spectrometry, the physical interaction partners of α-synuclein in rat primary cortical neurons were found to form a proteomic network that largely overlaps with the genetic network (Chung et al., Reference Chung, Khurana, Yi, Sahni, Loh, Auluck, Baru, Udeshi, Freyzon, Carr, Hill, Vidal, Ting and Lindquist2017). Altogether, the yeast α-synuclein models in combination with available genomic and proteomic tools have generated remarkable results in understanding the processes underlying PD.
Worm models
In agreement with the findings in yeast, several independent genetic screens in C. elegans found components of vesicle trafficking pathways to be involved in α-synuclein accumulation and toxicity. A genome-wide RNAi screen for suppressors of inclusion formation in the C. elegans model expressing α-synuclein in the body wall muscle cells revealed an enrichment of genes associated with the ER/Golgi and vesicle transport (van Ham et al., Reference van Ham, Thijssen, Breitling, Hofstra, Plasterk and Nollen2008). In a study targeting a subset of genes known or hypothesised to be involved in PD, two vesicle trafficking genes were discovered that protect against α-synuclein-mediated degeneration of dopaminergic neurons, in addition to known genetic PD factors such as DJ-1 and PINK1 (Hamamichi et al., Reference Hamamichi, Rivas, Knight, Cao, Caldwell and Caldwell2008). Finally, another screen performed on a pan-neuronal α-synuclein model led to the detection of several genes involved in endocytosis that protected against α-synuclein neurotoxicity, including two subunits of the adapter protein complex AP2 (Kuwahara et al., Reference Kuwahara, Koyama, Koyama, Yoshina, Ren, Kato, Mitani and Iwatsubo2008).
A gene that seemingly operates in a separate pathway that promotes aggregation was originally discovered in a screen targeting polyglutamine aggregation in the body wall muscle cells of C. elegans. This gene, moag-4 or SERF in humans, is similarly active in driving α-synuclein inclusion formation and toxicity, independently of known chaperone-dependent or insulin-signalling pathways (van Ham et al., Reference van Ham, Holmberg, van der Goot, Teuling, Garcia-Arencibia, Kim, Du, Thijssen, Wiersma, Burggraaff, van Bergeijk, van Rheenen, Jerre van Veluw, Hofstra, Rubinsztein and Nollen2010). Although the function of SERF in humans is not known at present, this gene may represent a suitable target for therapeutic intervention in protein misfolding diseases.
Fly models
Drosophila models of α-synuclein toxicity have been used less extensively in the context of unbiased-genetic screens. Using the reduced levels of dopamine in the heads of flies expressing α-synuclein A53T as a read-out, in combination with a genome-wide deletion library, the mitochondrial chaperone TRAP1 has been identified as a potent suppressor of toxicity (Butler et al., Reference Butler, Voigt, Lutz, Toegel, Gerhardt, Karsten, Falkenburger, Reinartz, Winklhofer and Schulz2012). Overexpressing TRAP1 in the α-synuclein A53T flies partially restores their climbing ability and resistance to oxidative stress, and the latter has been confirmed in rat neurons as well as in human cell lines, pointing towards a conserved link between α-synuclein pathology and mitochondrial dysfunction that is thought to play an important role in PD (Vicario et al., Reference Vicario, Cieri, Brini and Calì2018).
Rodent models
Several transcriptomics and proteomics studies have been performed in recent years in rodent models for PD based on the transgenic expression of α-synuclein. One investigation was carried out on a cross of an A53T α-synuclein overexpression model and a knock-out strain of the mitochondrial kinase PINK1, which causes an enhanced phenotype with ubiquitinated α-synuclein inclusions in the spinal cord and basal brain in mice from 1 year of age, and a reduced lifespan (Gispert et al., Reference Gispert, Brehm, Weil, Seidel, Rüb, Kern, Walter, Roeper and Auburger2015). Transcriptomic analysis of brain samples taken before the appearance of α-synuclein inclusions in this model has revealed perturbations in genes related to mitochondrial function, ubiquitin-mediated degradation, microtubule and vesicle trafficking, synaptic signalling and DNA-damage repair, compared to wild-type animals. At the protein level, analysis of ubiquitination patterns in 18-month-old mice has provided evidence for enhanced ubiquitination of α-synuclein itself upon the loss of PINK1, as well as reduced ubiquitination of the histone variant H2AX, a marker of sites of DNA damage (Gispert et al., Reference Gispert, Brehm, Weil, Seidel, Rüb, Kern, Walter, Roeper and Auburger2015).
A quantitative proteomics study on the model expressing α-synuclein with both A30P and A53T mutations has also highlighted the role of mitochondrial dysfunction and oxidative stress in the substantia nigra, as well as impairment of the ubiquitin–proteasome system, and ER stress (Yan et al., Reference Yan, Zhang, Jiao, Wang, He, Zhang, Zhang, Lv, Peng, Cai and Tian2017). A useful data source was furthermore established by a detailed analysis of the protein levels in distinct regions of the mouse brain, which has been performed for both non-transgenic mice and the line 61 PD mouse model (Jung et al., Reference Jung, Choi, Rousseaux, Malovannaya, Kim, Kutzera, Wang, Huang, Zhu, Maity, Zoghbi and Qin2017b). Comparison of the substantia nigra and an unaffected brain region in the PD model showed that membrane signalling proteins are upregulated specifically in the substantia nigra, whereas markers of extracellular matrix remodelling and inflammation are increased in aged PD mice compared to non-transgenic mice.
Further studies at the system level have mostly been performed on rodent models of PD induced by treatment with neurotoxins such as 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine, which have been remarkably successful in recapitulating phenotypic aspects of human PD (Tieu, Reference Tieu2011; Kasahara et al., Reference Kasahara, Choudhury, Nishikawa, Tanabe, Tsuji, Zhou, Ogawa, Yokoyama, Tanaka, Nomoto and Conn2017). Although these studies have identified perturbations in common pathways including oxidative phosphorylation, oxidative stress, apoptosis and synaptic transmission (Smith, Reference Smith2009), a comparative meta-analysis revealed that the gene expression signature of these models is relatively remote from that of human PD patients (Oerton and Bender, Reference Oerton and Bender2017). Overall, the current animal models of PD provide useful tools to study certain aspects of the multi-factorial disease process, such as the interplay between mitochondrial dysfunction and α-synuclein aggregation, but need to be improved to recapitulate better the complexity of the disease in the human brain.
Methodological challenges
An essential aim of in vivo models of human disease is to enable preclinical studies on systems that recapitulate the pathological processes observed in patients. In this section, we discuss four major challenges, two biophysical and two biological, that remain to be addressed in order to reach this goal:
(1) To develop longitudinal detection methods for protein aggregates. In AD and PD drug discovery pipelines, preclinical studies in mouse models often rely on the post-mortem quantification of protein deposits using methods based on immunohistochemistry or amyloid-sensitive fluorescent dyes (Sevigny et al., Reference Sevigny, Chiao, Bussière, Weinreb, Williams, Maier, Dunstan, Salloway, Chen, Ling, O'Gorman, Qian, Arastu, Li, Chollate, Brennan, Quintero-Monzon, Scannevin, Arnold, Engber, Rhodes, Ferrero, Hang, Mikulskis, Grimm, Hock, Nitsch and Sandrock2016; Drummond and Wisniewski, Reference Drummond and Wisniewski2017). Methods based on clearing techniques, including CLARITY (Chung and Deisseroth, Reference Chung and Deisseroth2013; Chung et al., Reference Chung, Wallace, Kim, Kalyanasundaram, Andalman, Davidson, Mirzabekov, Zalocusky, Mattis, Denisin, Pak, Bernstein, Ramakrishnan, Grosenick, Gradinaru and Deisseroth2013b), 3DISCO (Ertürk et al., Reference Ertürk, Becker, Jährling, Mauch, Hojer, Egen, Hellal, Bradke, Sheng and Dodt2012) and Scale (Hama et al., Reference Hama, Kurokawa, Kawano, Ando, Shimogori, Noda, Fukami, Sakaue-Sawano and Miyawaki2011) (Table 1), enable more quantitative assessments of the total amounts and distributions of these deposits, thereby offering more accurate endpoints on the anti-aggregation activities of therapeutic candidate compounds.
One may expect even better readouts, however, by the further development of methods to monitor longitudinally the inhibitory effects of these compounds. Approaches based on magnetic resonance imaging (MRI) and PET (Table 1) (Weiner et al., Reference Weiner, Veitch, Aisen, Beckett, Cairns, Green, Harvey, Jack, Jagust, Liu, Morris, Petersen, Saykin, Schmidt, Shaw, Shen, Siuciak, Soares, Toga and Trojanowski2013; Sevigny et al., Reference Sevigny, Chiao, Bussière, Weinreb, Williams, Maier, Dunstan, Salloway, Chen, Ling, O'Gorman, Qian, Arastu, Li, Chollate, Brennan, Quintero-Monzon, Scannevin, Arnold, Engber, Rhodes, Ferrero, Hang, Mikulskis, Grimm, Hock, Nitsch and Sandrock2016) already offer the possibility of carrying out this type of studies in mouse models. The development of PET tracers for α-synuclein will be particularly helpful in this direction (Merchant et al., Reference Merchant, Cedarbaum, Brundin, Dave, Eberling, Espay, Hutten, Javidnia, Luthman, Maetzler, Menalled, Reimer, Stoessl and Weiner2019).
To develop detection methods for protein oligomers. Both in AD and PD the most neurotoxic species are misfolded protein oligomers, and yet these species remain elusive and difficult to quantify, in particular in in vivo models (Table 1). Immunohistochemistry is typically used to probe the accumulation of Aβ, tau and α-synuclein in fixed samples, and conformation-specific antibodies are available for specific cases, including A11, which binds to β-sheet oligomers, and MC-1, which recognises misfolded tau. The recent introduction of enzyme-linked immunosorbent assay (ELISA)-based methods of greatly enhanced sensitivity, such as SIMOA (Kuhle et al., Reference Kuhle, Barro, Andreasson, Derfuss, Lindberg, Sandelius, Liman, Norgren, Blennow and Zetterberg2016), subject to further development, is opening novel opportunities in this direction. In our laboratory we have recently demonstrated the design of antibodies targeting specific epitopes (Sormanni et al., Reference Sormanni, Aprile and Vendruscolo2015; Aprile et al., Reference Aprile, Sormanni, Perni, Arosio, Linse, Knowles, Dobson and Vendruscolo2017, Aprile et al., in press), and this approach holds promise for discriminating different populations of oligomeric species based on the accessibility of a range of epitopes.
Advances in microscopy are furthermore expected to contribute to an improved molecular understanding of misfolded protein species in vivo. Super-resolution microscopy has revealed the morphology and the growth of fibrils at the nanometre scale in vitro and in cells (Kaminski Schierle et al., Reference Kaminski Schierle, Van De Linde, Erdelyi, Esbjörner, Klein, Rees, Bertoncini, Dobson, Sauer and Kaminski2011b; Duim et al., Reference Duim, Jiang, Shen, Frydman and Moerner2014; Pinotsi et al., Reference Pinotsi, Michel, Buell, Laine, Mahou, Dobson, Kaminski and Kaminski Schierle2016), and continuing efforts to implement these technologies in vivo have already allowed the use of RESOLFT and STED nanoscopy to visualise organelles and the cytoskeleton in C. elegans (Dreier et al., Reference Dreier, Castello, Coceano, Cáceres, Plastino, Vicidomini and Testa2019), Drosophila (Schnorrenberg et al., Reference Schnorrenberg, Grotjohann, Vorbrüggen, Herzig, Hell and Jakobs2016) and the mouse brain (Berning et al., Reference Berning, Willig, Steffens, Dibaj and Hell2012). Furthermore, the use of biosensors in combination with fluorescence microscopy may shed more light on the conformational state of protein aggregates in vivo, as demonstrated e.g. by the changes in the fluorescence lifetime of protein tags upon amyloid formation (Kaminski Schierle et al., Reference Kaminski Schierle, Bertoncini, Chan, van der Goot, Schwedler, Skepper, Schlachter, van Ham, Esposito, Kumita, Nollen, Dobson and Kaminski2011a; Laine et al., Reference Laine, Sinnige, Ma, Haack, Poudel, Gaida, Curry, Perni, Nollen, Dobson, Vendruscolo, Kaminski Schierle and Kaminski2019).
Another approach that holds promise for the detection of oligomeric protein species in animal models and patient material relies on seeded amplification in vitro. Several assays, e.g. protein misfolding cyclic amplification (PMCA) (Saborio et al., Reference Saborio, Permanne and Soto2001), the amyloid-seeding assay (Colby et al., Reference Colby, Zhang, Wang, Groth, Legname, Riesner and Prusiner2007) and real-time quaking-induced conversion (RT-QuIC) (Atarashi et al., Reference Atarashi, Satoh, Sano, Fuse, Yamaguchi, Ishibashi, Matsubara, Nakagaki, Yamanaka, Shirabe, Yamada, Mizusawa, Kitamoto, Klug, McGlade, Collins and Nishida2011) (Table 1), were originally designed to detect low concentrations of misfolded PrP molecules, based on the conversion of PrPC to PrPSc by the addition of seeds of the latter species. These assays have been adapted successfully to enable the detection of misfolded α-synuclein (Shahnawaz et al., Reference Shahnawaz, Tokuda, Waragai, Mendez, Ishii, Trenkwalder, Mollenhauer and Soto2017; Jung et al., Reference Jung, Lim, Bae, Lee, Choi, Lee, Lee, Kim and Lee2017a) and tau (Meyer et al., Reference Meyer, Dinkel, Rickman Hager and Margittai2014) in vitro, from mice and from human brain or cerebrospinal fluid. Further development of these approaches will undoubtedly help not only with the fundamental understanding of protein misfolding in vivo, but may also lead to diagnostic methods for patients.
(1) To create models to recapitulate the sporadic forms of AD and PD. The overexpression of Aβ, tau and α-synuclein or their mutational variants provides models that recapitulate the familial forms of AD and PD. In the sporadic forms, more subtle perturbations of the protein homoeostasis system are probably at play, which result in the aggregation of the disease-associated proteins starting from normal expression levels. The creation of specific animal models is becoming technically more feasible with developments in genetic-engineering techniques such as CRISPR/Cas9 (Knott and Doudna, Reference Knott and Doudna2018; Zhang, Reference Zhang2019), and by introducing additional genetic or environmental perturbations the need for the artificially high overexpression levels and non-physiological mutations on which current models often rely may be alleviated. For example, we have recently created a C. elegans model expressing Aβ42 at single-copy levels in only two neurons, which displays small perturbations in neuronal signalling and may be useful to study the onset of disease symptoms triggered by additional sources of cellular stress (Sinnige et al., Reference Sinnige, Ciryam, Casford, Dobson, De Bono and Vendruscolo2019).
(2) To create models to recapitulate the complexity of human AD and PD brain tissues. The in vivo models that we have described in this review have the inevitable limitation that they are genetically non-identical to humans, leading to differences in neuronal anatomy as well as processes such as ageing and the action of the immune system, which are critical in the study of neurodegenerative diseases. The use of iPSC-derived neurons and direct reprogramming of patient fibroblasts are promising approaches to circumventing some of these issues (Xu et al., Reference Xu, Du and Deng2015; Engle et al., Reference Engle, Blaha and Kleiman2018). In combination with 2D and 3D culturing methods, such as neuronal rosettes and brain organoids, respectively (Choi et al., Reference Choi, Kim, Hebisch, Sliwinski, Lee, D'Avanzo, Chen, Hooli, Asselin, Muffat, Klee, Zhang, Wainger, Peitz, Kovacs, Woolf, Wagner, Tanzi and Kim2014; Otani et al., Reference Otani, Marchetto, Gage, Simons and Livesey2016; Lancaster et al., Reference Lancaster, Corsini, Wolfinger, Gustafson, Phillips, Burkard, Otani, Livesey and Knoblich2017; Gonzalez et al., Reference Gonzalez, Armijo, Bravo-Alegria, Becerra-Calixto, Mays and Soto2018), these systems may increasingly provide avenues to elucidate disease mechanisms and to screen for therapeutics, in parallel to the use of traditional model organisms.
Conclusions
The use of in vivo models is instrumental in obtaining a detailed understanding of human diseases by connecting the observed phenotypes to the underlying molecular processes that give rise to them. In this review, we have described current in vivo models of AD and PD where the phenomenon of protein misfolding and aggregation plays a central role.
Studies based on these models have shown that mature amyloid fibrils or large aggregate inclusions are relatively inert and generally not associated with cytotoxicity, as they are limited in their ability to engage in aberrant interactions. By contrast, aggregation intermediates such as oligomeric species formed by Aβ, tau or α-synuclein are associated with the disturbance of cellular processes and with the onset and progress of neurodegenerative diseases. Some of the affected pathways appear to be disease-specific, as exemplified by the association of vesicle trafficking and mitochondrial function with the misfolding of α-synuclein, and of cytoskeletal transport and protein phosphorylation with that of tau. Other pathways may be perturbed more generally, most notably the protein homoeostasis network which becomes progressively impaired in ageing and neurodegeneration. Molecular chaperones have been shown to interact with all three proteins discussed in this paper, and to modulate their misfolding and aggregation (Sinnige et al., Reference Sinnige, Yu, Morimoto, Mendillo, Pincus and Scherz-Shouval2020).
However, the pathological mechanisms underlying these disorders are not yet fully understood, and no disease-modifying therapeutics have yet emerged. The creation of in vivo models that better recapitulate the sporadic forms of these diseases, as well as the development of biophysical methods for the accurate quantification of the aggregation process in living organisms, will be crucial in this regard.









 Open access
Open access