


INTRODUCTION
Cryptosporidiosis is the disease caused by the protozoan parasite Cryptosporidium, which affects many different species of mammals, including humans, birds, reptiles, amphibians and fish (Fayer, Reference Fayer2010; Chalmers and Katzer, Reference Chalmers and Katzer2013; Ryan et al. Reference Ryan, Fayer and Xiao2014). It has been identified as one of the most important causes of bovine neonatal enteritis worldwide (De Graaf et al. Reference De Graaf, Vanopdenbosch, Ortega-Mora, Abbassi and Peeters1999; Brook et al. Reference Brook, Hart, French and Christley2008; Silverlas et al. Reference Silverlas, Emanuelson, de and Bjorkman2009; Wyatt et al. Reference Wyatt, Riggs and Fayer2010).
The disease usually manifests itself as profuse watery diarrhoea, loss of appetite and abdominal pain (Clark and Sears, Reference Clark and Sears1996; Tzipori and Ward, Reference Tzipori and Ward2002; Klein et al. Reference Klein, Kleinova, Volek and Simunek2008) which lasts approximately 2 weeks with little or no long-term implications on animal health (Klein et al. Reference Klein, Kleinova, Volek and Simunek2008). Most animals will recover but in severe cases complications may arise and death can occur. There are currently over 27 recognized species of Cryptosporidium, four of which commonly infect cattle; Cryptosporidium parvum, Cryptosporidium andersoni, Cryptosporidium bovis and Cryptosporidium ryanae (Chalmers and Katzer, Reference Chalmers and Katzer2013; Ryan et al. Reference Ryan, Fayer and Xiao2014).
Detection of Cryptosporidium in farm livestock is usually done by microscopy (in veterinary diagnostic laboratories) or by molecular detection (research laboratories); of the two methods molecular detection is much more sensitive and allows full speciation (Chalmers and Katzer, Reference Chalmers and Katzer2013). One of the most common genes targeted for PCR detection of Cryptosporidium is the 18S rRNA gene, which is located on a multi-copy DNA element meaning that the PCR is more sensitive than those which target a single copy gene. The 18S rRNA gene is also one of the most commonly used genes for species discrimination and there are many 18S rRNA reference sequences available on the National Centre for Biotechnology Information website (www.ncbi.nlm.nih.gov). In order to identify species in Cryptosporidium-positive samples, it is necessary to carry out sequencing or PCR–RFLP (polymerase chain reaction followed by restriction fragment length polymorphism). These methods can be expensive, time consuming and it is very difficult to detect mixed infections as often only the most predominant species in a sample is detected (Cama et al. Reference Cama, Gilman, Vivar, Ticona, Ortega, Bern and Xiao2006). PCR–RFLP can also detect only a limited number of species.
A multiplex PCR test would enable the rapid speciation of a subset of Cryptosporidium species and previous attempts have been made to develop such a test (Patel et al. Reference Patel, Pedraza-Diaz and McLauchlin1999; Lindergard et al. Reference Lindergard, Nydam, Wade, Schaaf and Mohammed2003; Santin and Zarlenga, Reference Santin and Zarlenga2009); the most recent of which can detect the four most commonly found Cryptosporidium species in cattle (Santin and Zarlenga, Reference Santin and Zarlenga2009). This test cannot distinguish between C. bovis and C. ryanae and a further PCR amplification and sequencing would be required to distinguish these species.
In this paper, we describe the development of a multiplex PCR test to identify three cattle-adapted species (C. andersoni, C. bovis and C. ryanae) plus the zoonotic species C. parvum in a nested PCR. It is also possible with this multiplex PCR to detect other Cryptosporidium species by sequencing of the 18S rRNA gene, where none of the other species have been detected. This PCR test will be extremely useful in veterinary diagnostics as a tool for accurate diagnosis of scour in neonatal calves and as a research tool to investigate the epidemiology of cryptosporidiosis. The nssm-PCR would allow speciation but would not permit sub-typing, which would be necessary for outbreak investigation. Current diagnostic methods can only confirm that Cryptosporidium spp. are present in the feces, but cannot distinguish the disease causing C. parvum species from the less virulent C. andersoni which has been associated with a reduction in weight-gain and milk yield in adult cattle, C. bovis and C. ryanae, which have not yet been associated with the clinical disease. The test is already being used to monitor calves on a veterinary school farm to ensure that the calves used for practical classes are free from C. parvum to minimize the risk to the students.
MATERIALS AND METHODS
Design of Cryptosporidium species-specific primers
Alignments of multiple 18S rRNA gene sequences (between 50 and 100 for each species) for Cryptosporidium species (C. parvum, C. bovis, C. ryanae, C. andersoni, C. ubiquitum, C. hominis, C. muris, C. xiaoi and C. suis) were made using the BioEdit Software Program (www.mbio.ncsu.edu/bioedit/bioedit.html) to identify representative sequences for each species. Clustal W (www.ebi.ac.uk/Tools/msa/clustalw2) alignments of the representative sequences were made and regions for species-specific primers were identified which would produce amplicons of different sizes for each species when used with the 18S rRNA reverse primer (AL3032) (Xiao et al. Reference Xiao, Alderisio, Limor, Royer and Lal2000). The locations of the primers used are shown in Fig. 1; The outer primers are the same as those described by Xiao et al. Reference Xiao, Alderisio, Limor, Royer and Lal2000 (AL1691/AL1687) as are the first set of inner primers (AL1598 and AL3032) these amplify a 840 bp genus-specific fragment, the second inner primer set (CaF and AL3032) amplify a 625 bp fragment from C. andersoni; the third set (CrF and AL3032) amplify a 415 bp fragment from C. ryanae the forward primer in this set contains a degenerate base (R) where the C. ryanae sequence contains either an ‘A’ or a ‘G’; the fourth set (CphF and AL3032) amplify a 305 bp fragment from C. parvum, C. hominis and C. suis, which can be later distinguished with an additional PCR if desired and the final set of primers (CbF and AL3032) amplify a 241 bp fragment from C. bovis. The primer sequences are shown in Table 1.

Fig. 1. Positions of species-specific primers on the 18S rRNA gene plus the location of the 18S rRNA internal primers.
Table 1. Primer sequences, size of amplicon and species detected with each primer pair

a The degenerate base in the C. ryanae primer is shown in bold and underlined.
Fecal sample collection and DNA extraction
Fecal samples (n = 498) were collected from 190 calves and 308 adult cattle from 21 beef farms as part of another study. These samples were collected from calves aged 7–14 days regardless of clinical signs (n = 116), all calves suffering from diarrhoea regardless of age (n = 74), adult cattle pre- and post-calving (n = 177 and 120, respectively) and other adult cows (n = 11). Samples were collected by the farmers and sent to the Moredun Research Institute by their veterinary practices.
DNA was extracted from the calf feces processing with a QIAamp DNA Stool Minikit (Qiagen) according to the manufacturer's instructions except that 10× freeze–thaw cycles in liquid nitrogen were carried out after adding 200 µg sample to lysis buffer (provided in the kit). Adult feces were subjected to a sedimentation step prior to DNA extraction. The resultant pellet was added to lysis buffer and subjected to 10× freeze–thaw cycles in liquid nitrogen followed by processing by QIAamp DNA Stool Minikit (Qiagen) according to the manufacturer's instructions.
To identify Cryptosporidium-positive samples 18S nested PCR was carried out as described by Xiao et al. Reference Xiao, Alderisio, Limor, Royer and Lal2000. This work was carried out as part of another study at the Moredun Research Institute. Microscopic examination of the feces was not carried out for any of the samples.
Testing of individual species-specific primers
First round reactions were carried out as described by Xiao et al. Reference Xiao, Alderisio, Limor, Royer and Lal2000 using DNA from known positive samples for each of the four species. Secondary reactions were carried out to test each individual species-specific forward primer with the reverse 18S rRNA primer (AL3032). First round PCR products were diluted with 50 µL dH2O and 1 µL of the dilution used as a template in a reaction containing 2·5 µL 10× PCR buffer (45 mm Tris–HCL pH 8·8, 11 mm (NH4)2SO4, 4·5 mm MgCl2, 4·4 µ m EDTA, 113 µg mL−1 BSA, 1 mm of each of the four deoxyribonucleotide triphosphates) (Katzer et al. Reference Katzer, Canton, Burrells, Palarea-Albaladejo, Horton, Bartley, Pang, Chianini, Innes and Benavides2014), 0·5 unit BioTaq (Bioline, UK) and 10 µ m forward and reverse primers. The final volume of each reaction was made up to 25 µL using dH2O.
Cycling conditions were 3 min at 94 °C, followed by 35 cycles of 45 s at 94 °C, 45 s on a temperature gradient of 49–59 °C and 1 min at 72 °C. The final extension was 7 min at 72 °C. Second round PCR products were visualized following electrophoresis on a 1·5% agarose gel stained with GelRed™ (Biotium, UK) on an AlphaImager 2200.
Field samples
Primers were tested on a panel of the only available DNA samples, which included known positive samples for C. parvum, C. andersoni, C. bovis and C. ryanae from naturally infected cattle aged from 1 day old to adult animals (n = 128). Results from the nssm-PCR were compared with sequence data for all of these samples.
Sequence analysis
To confirm the results of the nssm-PCR when testing field samples, sequence analysis of the 18S rRNA amplicon was carried out. Sequencing of PCR products was carried out by GATC (GATC, Germany) using their ‘Light-run’ service. Roughly 5 μL of purified PCR product was sent at a concentration of between 20 and 80 ng µL with 5 µL of the appropriate primer at a concentration of 5 pm µL. In some circumstances, instead of the light-run service, unpurified PCR products were sent to GATC from sequencing using their ‘Supremerun96’ sequencing option which includes PCR purification. With this service 20 µL PCR product was sent with a separate 1·5 mL tube containing 50 µL of the appropriate primer at a concentration of 10 pm µL. Sequences were analysed using BioEdit Sequence Alignment Software (www.mbio.ncsu.edu/bioedit/bioedit.html) and Chromas Lite (www.technelysium.com).
Sensitivity
The sensitivity of the nssm-PCR assay was assessed as part of another study (Wells B, Thomson S, Innes E.A and Katzer F Development of a sensitive method to extract Cryptosporidium oocysts from adult cattle fecal samples – submitted). Fecal samples were spiked with C. parvum oocysts, subjected to different concentration steps and DNA extracted. The DNA was then amplified using the nssm-PCR.
RESULTS
Primer design
The primer designed for C. parvum will also amplify C. hominis and C. suis, the C. andersoni primer will also amplify C. muris and the C. bovis primer will amplify C. xiaoi as the 18S region of these species are very similar (Fig. 1). Cryptosporidium suis and C. hominis have only been found very rarely in cattle (Smith et al. Reference Smith, Nichols, Mallon, MacLeod, Tait, Reilly, Browning, Gray, Reid and Wastling2005; Fayer et al. Reference Fayer, Santin, Trout and Greiner2006) and C. muris and C. xiaoi have yet not been identified in cattle.
Cryptosporidium-specific 18S rRNA PCR
All 498 samples were tested for the presence of Cryptosporidium parasite DNA using the 18S genus-specific PCR, 4·13% (n = 13) of the samples from adult cattle and 60·5% (n = 115) of the samples from calves tested positive for Cryptosporidium. These samples were used to test the nssm-PCR.
Individual species PCR
Amplicons of the expected size for each species (C. andersoni, C. ryanae, C. bovis and C. parvum) were obtained when tested in a single species PCR with the species-specific forward primer and the 18S rRNA reverse primer. DNA was obtained from samples (confirmed by sequencing) from calves aged <1 week (C. parvum DNA), calves aged 3 months (C. bovis and C. ryanae DNA) and an adult cow (C. andersoni DNA).
Nested species-specific multiplex PCR
Amplicons of the expected size for each of the four species (C. andersoni, C. ryanae, C. parvum and C. bovis – individually and mixed combinations) were obtained using the nssm-PCR assay (Fig. 2) from a variety of cattle samples collected from calves aged 7–14 days regardless of clinical signs (n = 116), all calves suffering from diarrhoea regardless of age (n = 74), adult cattle pre- and post-calving (n = 177 and n = 120, respectively) and other adult cows (n = 11). The expected amplicons for all four cattle species [C. parvum (n = 92), C. andersoni (n = 1), C. ryanae (only in mixed infections) and C. bovis (n = 13)] were observed as were various combinations of mixed infections (Fig. 2).
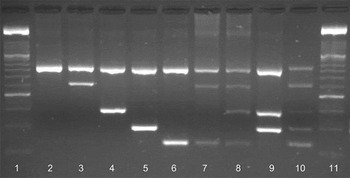
Fig. 2. Amplification of Cryptosporidium spp. on a 2% agarose gel stained with GelRedTM (Biotum, UK). Lane 1, 100 bp marker XIV (Roche); lane 2, 18S rRNA genus-specific amplicon (840 bp); lane 3, C. andersoni (625 bp); lane 4, C. ryanae (415 bp); lane 5, C. parvum (305 bp); lane 6, C. bovis (241 bp); lane 7, C. andersoni and C. bovis; lane 8, C. andersoni, C. ryanae and C. bovis; lane 9, C. ryanae and C. parvum; lane 10, nssm-PCR molecular marker; lane 11, 100 bp marker XIV (Roche). Amplicons in lanes 2–6 were produced using cloned DNA and lanes 7–9 were from naturally infected cattle.
Occasionally an additional non-specific band appeared with the amplicon for C. parvum but of a different size to the species-specific bands. The results of the nssm-PCR were compared with sequence data for the 18S rRNA gene of these samples. In all cases, the single species of Cryptosporidium identified by nssm-PCR matched the species identified by sequencing. In addition, the nssm-PCR identified mixed infections in 14 of the samples examined. The mixed infections included; C. bovis, C. parvum and C. ryanae (n = 1), C. ryanae and C. bovis (n = 5), C. parvum and C. bovis (n = 1), C. parvum and C. ryanae (n = 1) and C. andersoni and C. ryanae (n = 6). These mixed infections were not identified by sequencing. Eight of the samples failed to amplify using the nssm-PCR.
Sensitivity
The sensitivity of the nssm-PCR when tested on fecal samples spiked with C. parvum oocysts was 5 opg (oocysts per gram of feces) (Table 2).
Table 2. Sensitivity of nssm-PCR when tested on DNA extracted from feces spiked with 0, 5, 10, 100 or 1000 C. parvum oocysts per gram
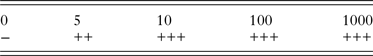
−, negative; +, positive 1/3 PCRs; ++, positive 2/3 PCRs; +++, positive 3/3 PCRs.
DISCUSSION
In this paper, we describe the development of a multiplex PCR test, which allows the discrimination of four of the most commonly detected Cryptosporidium species of cattle (one of which poses a risk to human health). The test allows the differentiation of these species by generating distinct amplicons of the 18S rRNA gene: C. andersoni (625 bp), C. ryanae (415 bp), C. parvum (305 bp) and C. bovis (241 bp) in addition to a Cryptosporidium genus-specific amplicon (840 bp). Currently, the most popular method of species identification in Cryptosporidium-positive samples is gene sequencing of the 18S rRNA gene. This method is very sensitive and can detect a large range of Cryptosporidium species and genotypes, but is limited when mixed infections occur as it has been shown that in a mixed sample either the dominant Cryptosporidium species is preferentially amplified by PCR meaning that only one species will be identifiable by sequencing or the sequence is unreadable (Santin et al. Reference Santin, Trout, Xiao, Zhou, Greiner and Fayer2004; Cama et al. Reference Cama, Gilman, Vivar, Ticona, Ortega, Bern and Xiao2006; Silverlas et al. Reference Silverlas, Naslund, Bjorkman and Mattsson2010). PCR amplification followed by sequence analysis is also more costly and time-consuming than multiplex PCR. The multiplex PCR described here allows the detection of mixed infections of up to four different common cattle species of Cryptosporidium and is an excellent method for detecting underlying infections of zoonotic C. parvum in calves which may have higher burdens of the less pathogenic Cryptosporidium species. This has very important implications for human health and the new test has also proven useful when determining which calves are most suitable for use in calf handing classes at a local veterinary school to reduce the risk of Cryptosporidium infection to students. It has also been used to identify the species of Cryptosporidium present in feces from adult cattle and their calves as part of a whole catchment study in the North East of Scotland (Wells et al. Reference Wells, Shaw, Hotchkiss, Gilray, Ayton, Green, Katzer, Wells and Innes2015).
In addition to the easy and quick visual differentiation of the four common cattle species other species of Cryptosporidium may be identified by sequencing of the 18S rRNA gene when a fragment responding to the Cryptosporidium genus-specific amplicon (840 bp) is amplified with no species-specific amplicons. At present, using this method, it is not possible to distinguish between C. parvum and C. hominis however, as C. hominis has rarely been detected in cattle (Smith et al. Reference Smith, Nichols, Mallon, MacLeod, Tait, Reilly, Browning, Gray, Reid and Wastling2005; Park et al. Reference Park, Guk, Han, Shin, Kim and Chai2006) there is, at this stage, no real need to distinguish between these two species.
The advantages of the PCR protocol described here over other methods used for species discrimination (PCR–RFLP, sequencing) are: (1) the ability to detect mixed infections, which may be missed by other methods; (2) the ability to identify other Cryptosporidium species by further sequencing when none of the ‘cattle species’ are detected as other species of Cryptosporidium may be a risk to other animals or humans; (3) the ability to test large numbers of samples quickly and easily; and (4) the ability to detect subclinical infections of the zoonotic species C. parvum in animals shedding high numbers of the non-zoonotic species.
FINANCIAL SUPPORT
The authors would like to thank AHDB, EBLEX, HCC, QMS, DairyCo, BBSRC for funding awarded to Sarah Thomson and The Scottish Government for funding awarded to Elisabeth Innes and Frank Katzer.





 Open access
Open access