



Introduction
Viruses cause significant illness and death in humans, animals, and plants. A detailed mechanistic understanding of virus structure, viral protein dynamics and function, host interactions, and viral replication strategies is critically important for the development of diverse anti-viral strategies. Much of our current understanding of the replication cycle of viruses has been compiled over several decades using a variety of structural biology and biochemical methods, with significant contributions from the fields of electron microscopy and X-ray crystallography. More recently, however, single-molecule biophysical approaches such as single-particle tracking, super-resolution microscopy, fluorescence correlation spectroscopy, single-molecule fluorescence in situ hybridisation (smFISH), and single-molecule fluorescence (or Förster) resonance energy transfer (smFRET) have become an important method of choice with which to address many fundamental questions in virology.
Since its first implementation in 1996 (Ha et al., Reference Ha, Ogletree, Chemla, Selvin, Weiss and Weiss1996), smFRET has been widely used to provide new insights into many fundamental biological processes, such as DNA maintenance and repair, protein conformation dynamics, ion channel dynamics, receptor–ligand interactions, nucleic acid structure and conformation, and membrane transport (Lerner et al., Reference Lerner, Cordes, Ingargiola, Alhadid, Chung, Michalet and Weiss2018). Unlike ensemble biochemical and structural methods that report on the mean properties of billions of molecules, thus averaging the measured parameters over the entire molecular population, smFRET offers a way of disentangling functionally important heterogeneity. It allows the real-time observation of transient intermediates, interconverting states, molecular interactions, and importantly, the study of dynamic processes (Figure 1a).

Figure 1. The theory of smFRET. a) A biomolecule that switches between two conformations. A short distance between a donor (green) and acceptor (red) fluorophore gives rise to high FRET, while little or no FRET is observed when the fluorophores are far apart. b) The relationship between the inter-fluorophore distance, R, and the FRET efficiency, E. The Förster radius R 0 refers to the distance at which the FRET efficiency is 50%. c) A schematic of confocal smFRET measurements on freely diffusing molecules. d) Example histogram of confocal data generated by plotting the FRET efficiency versus the stoichiometry for each fluorescent burst of photons. Stoichiometry is calculated using alternating-laser excitation (Kapanidis et al., Reference Kapanidis, Lee, Laurence, Doose, Margeat and Weiss2004), which allows identification of doubly labelled molecules. e) A schematic of total internal reflection fluorescence (TIRF) smFRET measurements on immobilised molecules. When light strikes the glass-water interface at a sufficiently high angle - greater than the critical angle (Ɵc) - it is totally internally reflected. When the angle of incidence is greater than the critical angle light in totally internally reflected. f) Example time trace showing FRET from an immobilised molecule. Clear anti-correlations in donor–acceptor fluorescence are characteristic of FRET dynamics.
FRET is often described as a ‘spectroscopic ruler’ (Stryer and Haugland, Reference Stryer and Haugland1967) – the result of a distance-dependent and non-radiative energy transfer from a high-energy molecule (donor) to a lower-energy molecule (acceptor). The energy transfer between donor and acceptor molecules is dependent on their proximity, as described by the Förster theory (Förster, Reference Förster1948) (Figure 1b). Förster showed that the rate of excitation energy transfer between a suitable donor and acceptor fluorophore is proportional to the inverse 6th power of the distance separating them:
 $$ E\hskip0.35em =\hskip0.35em 1/\Big(1+{\left(R/{R}_0\right)}^6, $$
$$ E\hskip0.35em =\hskip0.35em 1/\Big(1+{\left(R/{R}_0\right)}^6, $$
where E is the FRET efficiency, R is the inter-fluorophore distance, and R 0 is the Förster radius, a proportionality constant that depends on the interaction between the transition dipoles of donor and acceptor and represents the distance between the donor and the acceptor at which energy transfer is 50%. The range of distances that can be accurately measured with FRET is ~3 to 9 nm, making it well suited to measure the dimensions and distance dynamics of viral proteins.
To experimentally realise single-molecule FRET, it is necessary to detect single fluorescent molecules against the background created by a vast excess of solute molecules. To achieve this, the molecules of interest are labelled with specific donor and acceptor fluorophores, which are ideally stable under high photon flux, have high molar absorptivity and fluorescence quantum yield, and undergo minimal blinking (Sasmal et al., Reference Sasmal, Pulido, Kasal and Huang2016). Such fluorophores are generally small (~1 nm) organic molecules that are usually attached via flexible linkers to defined positions on the protein(s) under investigation. Common dyes include the cyanine dyes Cy3 and Cy5, as well as the Alexa Fluor and ATTO dye series. The positioning of fluorophores on the protein of interest should be carefully chosen such that the distance between a donor and an acceptor is within the Förster radius (Lerner et al., Reference Lerner, Barth, Hendrix, Ambrose, Birkedal, Blanchard, Börner, Sung Chung, Cordes, Craggs, Deniz, Diao, Fei, Gonzalez, Gopich, Ha, Hanke, Haran, Hatzakis, Hohng, Hong, Hugel, Ingargiola, Joo, Kapanidis, Kim, Laurence, Lee, Lee, Lemke, Margeat, Michaelis, Michalet, Myong, Nettels, Peulen, Ploetz, Razvag, Robb, Schuler, Soleimaninejad, Tang, Vafabakhsh, Lamb, Seidel and Weiss2021).
The two most popular approaches for visualising labelled molecules are confocal microscopy and total internal reflection fluorescence (TIRF) microscopy. While both approaches ensure a high signal-to-noise ratio and relatively high throughput, this is achieved in different ways. In diffusion-based confocal microscopy, single-molecule detection is achieved by focusing a laser beam into a freely diffusing solution of labelled molecules using a high numerical aperture objective lens (Figure 1c). The small size of the resulting excitation volume (~1 fL), together with a confocal pinhole to remove out-of-focus light, provides the required spatial selection so that only fluorescence photons resulting from the FRET process are detected (Schuler, Reference Schuler2013). The photons are focused on highly sensitive detectors, typically avalanche photodiodes (APDs), which provide a time resolution in the 50 ps range. Each transit of a labelled molecule through the confocal volume will lead to the detection of a fluorescent ‘burst’ of photons, which are then used to determine FRET efficiencies, providing crucial information on protein conformations (Figure 1d). The advantage of imaging in solution is the low concentration of fluorescently labelled molecules (~10 to 100 pM), resulting in a negligible probability of two molecules residing in the confocal volume at the same time (Schuler, Reference Schuler2013).
Alternatively, labelled molecules are immobilised on a surface and visualised using TIRF microscopy, an optical technique that excites and images fluorophores in an extremely thin axial region. The method is based on the principle that when excitation light is totally internally reflected at the sample-coverglass interface, an electromagnetic field, called the evanescent wave, is generated in the liquid at the same frequency as the excitation light (Fish, Reference Fish2009). Since the intensity of the evanescent wave exponentially decays with distance from the surface of the glass, only fluorescent molecules within a few hundred nanometres of the coverglass are efficiently excited (Fish, Reference Fish2009) (Figure 1e). Total internal reflection of incident light is achieved either by illuminating the sample through an objective or through a quartz prism. Using TIRF, hundreds to thousands of dye-labelled molecules can be imaged simultaneously in one field of view, allowing individual protein dynamics to be recorded until the fluorophores bleach (typically within seconds to minutes) (Figure 1f). It typically has a temporal resolution of a few tens of milliseconds, but this is improving with technological advances (Lerner et al., Reference Lerner, Barth, Hendrix, Ambrose, Birkedal, Blanchard, Börner, Sung Chung, Cordes, Craggs, Deniz, Diao, Fei, Gonzalez, Gopich, Ha, Hanke, Haran, Hatzakis, Hohng, Hong, Hugel, Ingargiola, Joo, Kapanidis, Kim, Laurence, Lee, Lee, Lemke, Margeat, Michaelis, Michalet, Myong, Nettels, Peulen, Ploetz, Razvag, Robb, Schuler, Soleimaninejad, Tang, Vafabakhsh, Lamb, Seidel and Weiss2021).
In this review, we discuss the application of smFRET to the study of a wide number of disease-causing human viruses. In particular, we consider the use of smFRET to study i) viral glycoprotein dynamics in HIV-1, influenza, Ebola, and SARS-CoV-2; ii) viral helicases in the hepatitis C virus and vaccinia virus; iii) viral proteins involved in HIV reverse transcription; and iv) the influenza RNA-dependent RNA polymerase. We review the key contributions of smFRET to the study of viral protein and nucleic acid conformational dynamics, highlighting the crucial role that these studies have played in furthering our understanding of these important pathogens.
Conformational dynamics of viral surface glycoproteins measured by smFRET
Many viruses are coated in glycoproteins, including the HIV-1 envelope (Env), influenza haemagglutinin (HA), Ebola glycoprotein (GP), and the SARS-CoV-2 spike (S). Viral surface glycoproteins are responsible for binding to the host-cell surface and fusing with the membrane in order to release the viral capsid or genome into the cell cytoplasm. The glycoproteins are usually trimers, where each protomer consists of a transmembrane fusion domain and a surface-facing subunit (Figure 2). Upon surface subunit binding to the target cell receptor, the fusion subunit exposes a fusion loop and initiates membrane fusion (Belouzard et al., Reference Belouzard, Millet, Licitra and Whittaker2012; Shang et al., Reference Shang, Luo, Ye, Geng, Auerbach, Li and Li2020a). Viral glycoprotein structure and fusion mechanisms have been extensively explored by X-ray crystallography and cryo-electron microscopy (cryo-EM), which have revealed single-molecule detail by allowing the alignment of large EM datasets of two-dimensional projections to produce densities in three dimensions (cryo-EM single-particle reconstruction (SPR)). It is also possible to determine the potential flexibility of structures by tilting and aligning sample volumes of particles in situ (cryo-electron tomography (cryo-ET) and subtomogram averaging) (Wan and Briggs, Reference Wan and Briggs2016). Coupled with molecular dynamics (MD) simulations, conformational dynamics and transitions between static structures can now even be predicted from these structures. However, due to the stringent sample preparation conditions of cryo-EM and limitations in field of view and labelling in aligned SPR or cryo-ET structures (Zhang, Reference Zhang2013), it is difficult to capture fusion dynamics between receptor–ligand interactions and the rapid fluctuations that occur between conformational states (Lee et al., Reference Lee, Ozorowski and Ward2016; Ward and Wilson, Reference Ward and Wilson2017). The labelling and real-time capability of smFRET have provided valuable insight into the transitions between conformational structures identified by EM and temporal dynamics of viral glycoproteins before, during, and after receptor binding and fusion. Viral surface glycoproteins are major targets of immune cells and neutralising antibodies and are often responsible for immune escape (Belouzard et al., Reference Belouzard, Millet, Licitra and Whittaker2012); therefore, identifying surface structures and transitions between them could elucidate fusion mechanisms, immune evasion strategies, and binding partners useful for new anti-viral vaccines or therapeutics (Sasmal et al., Reference Sasmal, Pulido, Kasal and Huang2016).

Figure 2. Viral glycoproteins have a common architecture. Viral surface glycoproteins often have similar trimer structures. Each protomer consists of a surface facing binding domain and a transmembrane fusion subunit. a) HIV-1 Env contains 3 protomers of gp120 (receptor-binding subunit) and gp41 (transmembrane subunit); left: crystal structure of BG505 SOSIP gp140 mimic of the Env ectodomain (pdb: 4NCO)16 where each protomer is distinguished by shades of purple, right: schematic. b) The full crystalised H5N1 influenza HA and schematic illustrate a trimer of HA1 and HA2 domains (pdb: 2FK0) (Stevens et al., Reference Stevens, Tumpey, Taubenberger, Paulson, Wilson and Wilson2006). c) The ectodomain of the Ebola GP without the mucin-like domain consists of a trimer of heterodimers GP1 and GP2 (pdb: 7SWD) (Milligan et al., Reference Milligan, Davis, Yu, Ilinykh, Huang, Halfmann, Cross, Borisevich, Agans, Geisbert, Chennareddy, Goff, Piper, Hui, Shaffer, Buck, Heinrich, Branco, Crozier, Holbrook, Kuhn, Kawaoka, Glass, Bukreyev, Geisbert, Worwa, Ahmed and Saphire2022). d) The SARS-CoV-2 Spike protein also contains a trimer of S1 (receptor-binding domain) and S2 (fusion) subunits (pdb: 6VYB) (Walls et al., Reference Walls, Park, Tortorici, Wall, AT and Veesler2020).
The HIV-1 envelope protein
The HIV-1 Env glycoprotein is responsible for binding to the T cell CD4 receptor and the CCR5 or CXCR4 coreceptors during viral entry (Lu, Reference Lu2021). The Env precursor gp160 is cleaved by furin proteases to form gp120, which binds to target receptors, and gp41, the fusion subunit (Lee et al., Reference Lee, Ozorowski and Ward2016; Ward and Wilson, Reference Ward and Wilson2017). Mature Env, a trimer of gp120/gp41 heterodimers, undergoes a series of structural rearrangements in both subunits upon engaging with CD4 and the coreceptors. CD4-binding induces conformational changes in gp120 to expose coreceptor-binding sites, whereas CCR5 or CXCR4 binding to gp120 is thought to activate refolding events in gp41, eventually leading to viral membrane fusion. smFRET has been used to characterise the conformational changes in gp120 during target receptor binding, the findings of which have been summarised in a number of excellent reviews (Howard and Munro, Reference Howard and Munro2019; Lu et al., Reference Lu, Ma and Mothes2019b; Lu, Reference Lu2021).
Munro et al. (Reference Munro, Gorman, Ma, Zhou, Arthos, Burton, Koff, Courter, Smith, Kwong, Blanchard and Mothes2014) assessed the movement of variable loops in gp120 during receptor binding by labelling the V1 loop with a donor fluorophore, and the V4 or V5 loops with an acceptor fluorophore, followed by smFRET analysis via TIRF microscopy (Figure 1). The transfer efficiency between fluorophores was used to determine the molecular distances between the loops at different stages of receptor activation. Unliganded Env dynamically sampled three conformational states, in which ‘state 1’ (a low-FRET closed state) was predominant (Munro et al., Reference Munro, Gorman, Ma, Zhou, Arthos, Burton, Koff, Courter, Smith, Kwong, Blanchard and Mothes2014). In the presence of soluble CD4, the FRET state increased to a high-FRET state (0.6) and decreased to an intermediate-FRET (0.3) when a coreceptor mimic was introduced. This suggests that the Env glycoprotein undergoes three distinct conformational changes: pre-receptor activated Env (state 1); a default intermediate (state 2), in which a single CD4 is engaged with the trimer; and a stabilised open Env where all three protomers are bound to CD4 (state 3) (Figure 3a) (Munro et al., Reference Munro, Gorman, Ma, Zhou, Arthos, Burton, Koff, Courter, Smith, Kwong, Blanchard and Mothes2014).
Alsahafi et al. have suggested a fourth state (‘state 2A’), an off-path semi-open conformation that is highly vulnerable to antibody-dependent cellular cytotoxicity (ADCC), a mechanism of cell-mediated immune defence whereby an immune cell lyses a target cell whose membrane-surface antigens have been bound by specific antibodies (Figure 3b). ADCC has been previously associated with protection against HIV during the RV144 vaccine trial (Prévost et al., Reference Prévost, Zoubchenok, Richard, Veillette, Pacheco, Coutu, Brassard, Parsons, Ruxrungtham, Bunupuradah, Tovanabutra, Hwang, Moody, Haynes, Bonsignori, Sodroski, Kaufmann, Shaw, Chenine and Finzi2017; Alsahafi et al., Reference Alsahafi, Kazemi, Richard, Ding, Bhattacharyya, das, Anand, Prévost, Tolbert, Lu, Medjahed, Gendron-Lepage, Ortega Delgado, Kirk, Melillo, Mothes, Sodroski, Smith, Kaufmann, Wu, Pazgier, Rouiller, Finzi, Munro and Munro2019). If ADCC indeed works by specific clearance of open states of Env, this has the potential to be exploited therapeutically by forcing Env into open conformations using CD4 mimetics (Alsahafi et al., Reference Alsahafi, Kazemi, Richard, Ding, Bhattacharyya, das, Anand, Prévost, Tolbert, Lu, Medjahed, Gendron-Lepage, Ortega Delgado, Kirk, Melillo, Mothes, Sodroski, Smith, Kaufmann, Wu, Pazgier, Rouiller, Finzi, Munro and Munro2019). smFRET imaging has also provided additional mechanistic understandings of how the Env trimer responds to anti-viral inhibitors, for example, the entry inhibitor Temsavir was shown to reduce the occupancy of downstream open conformations and stabilise Env in the closed state 1 (Alsahafi et al., Reference Alsahafi, Kazemi, Richard, Ding, Bhattacharyya, das, Anand, Prévost, Tolbert, Lu, Medjahed, Gendron-Lepage, Ortega Delgado, Kirk, Melillo, Mothes, Sodroski, Smith, Kaufmann, Wu, Pazgier, Rouiller, Finzi, Munro and Munro2019; Bruel and Schwartz, Reference Bruel and Schwartz2019; Lu et al., Reference Lu, Ma, Castillo-Menendez, Gorman, Alsahafi, Ermel, Terry, Chambers, Peng, Zhang, Zhou, Reichard, Wang, Grover, Carman, Gardner, Nikić-Spiegel, Sugawara, Arthos, Lemke, Smith, Farzan, Abrams, Munro, McDermott, Finzi, Kwong, Blanchard, Sodroski and Mothes2019a).

Figure 3. smFRET analysis reveals conformational states and dynamics of HIV-1 Env. a) Model of Env-mediated membrane fusion. ‘State 1’ (a low-FRET closed state) is thought to represent unliganded Env, while CD4 binding induces a partially open high-FRET conformation (‘state 2’), followed by coreceptor binding which results in the transition to a stabilised open Env, where all 3 protomers are bound to CD4 (intermediate-FRET ‘state 3’). Following CD4 activation fusion of the viral and cellular membranes occurs, leading to virus entry. b) State 1 can also transition into a fourth state (‘state 2A’), representing an off-path semi-open conformation that is highly vulnerable to antibody-dependent cellular cytotoxicity (ADCC). Adapted from (Bruel and Schwartz, Reference Bruel and Schwartz2019).
Infection with HIV-1 results in targeting by anti-Env antibodies. smFRET has been used to show that many broadly neutralising antibodies bind preferentially to closed pre-fusion state 1 Env (Alsahafi et al., Reference Alsahafi, Kazemi, Richard, Ding, Bhattacharyya, das, Anand, Prévost, Tolbert, Lu, Medjahed, Gendron-Lepage, Ortega Delgado, Kirk, Melillo, Mothes, Sodroski, Smith, Kaufmann, Wu, Pazgier, Rouiller, Finzi, Munro and Munro2019; Bruel and Schwartz, Reference Bruel and Schwartz2019; Lu et al., Reference Lu, Ma, Castillo-Menendez, Gorman, Alsahafi, Ermel, Terry, Chambers, Peng, Zhang, Zhou, Reichard, Wang, Grover, Carman, Gardner, Nikić-Spiegel, Sugawara, Arthos, Lemke, Smith, Farzan, Abrams, Munro, McDermott, Finzi, Kwong, Blanchard, Sodroski and Mothes2019a), whereas non-neutralising antibodies stabilise the open state 3 (Bruel and Schwartz, Reference Bruel and Schwartz2019; Lu et al., Reference Lu, Ma, Castillo-Menendez, Gorman, Alsahafi, Ermel, Terry, Chambers, Peng, Zhang, Zhou, Reichard, Wang, Grover, Carman, Gardner, Nikić-Spiegel, Sugawara, Arthos, Lemke, Smith, Farzan, Abrams, Munro, McDermott, Finzi, Kwong, Blanchard, Sodroski and Mothes2019a). This has significant implications for the design of Env-mimicking vaccines that aim to induce broadly neutralising antibodies but not non-neutralising antibodies (Lee et al., Reference Lee, Ozorowski and Ward2016). One of the most promising Env-based HIV-1 vaccine candidates is a recombinant, truncated gp140 trimer called SOSIP.664 (Sanders et al., Reference Sanders, Derking, Cupo, Julien, Yasmeen, de Val, Kim, Blattner, de la Peña, Korzun, Golabek, de los Reyes, Ketas, van Gils, King, Wilson, Ward, Klasse and Moore2013; Lee et al., Reference Lee, Ozorowski and Ward2016). Previously, numerous high-resolution structures of pre-fusion Env revealed a similar architecture, assumed to resemble the primary conformation of virus-embedded native Env in ‘state 1’ (Julien et al., Reference Julien, Cupo, Sok, Stanfield, Lyumkis, Deller, Klasse, Burton, Sanders, Moore, Ward and Wilson2013; Sanders et al., Reference Sanders, Derking, Cupo, Julien, Yasmeen, de Val, Kim, Blattner, de la Peña, Korzun, Golabek, de los Reyes, Ketas, van Gils, King, Wilson, Ward, Klasse and Moore2013; Lee et al., Reference Lee, Ozorowski and Ward2016). smFRET experiments comparing SOSIP mutants and wild-type Env unexpectedly showed that SOSIP.664 resembled the default intermediate, state 2, not state 1 (Bruel and Schwartz, Reference Bruel and Schwartz2019; Lu et al., Reference Lu, Ma, Castillo-Menendez, Gorman, Alsahafi, Ermel, Terry, Chambers, Peng, Zhang, Zhou, Reichard, Wang, Grover, Carman, Gardner, Nikić-Spiegel, Sugawara, Arthos, Lemke, Smith, Farzan, Abrams, Munro, McDermott, Finzi, Kwong, Blanchard, Sodroski and Mothes2019a), suggesting that vaccine candidates based on SOSIP.664 designs may not induce a broadly neutralising antibody response. The structure of the conformational state behind the State 1 observed by smFRET remains unknown. Munro et al. (Reference Munro, Gorman, Ma, Zhou, Arthos, Burton, Koff, Courter, Smith, Kwong, Blanchard and Mothes2014) also showed the strain of HIV-1 influenced the FRET efficiency, where the neutralisation-resistant HIV-1 JR-FL transitioned between FRET states, especially the intermediate-FRET state, much less frequently. This suggests the neutralisation-resistant HIV-1 JR-FL spends more time in the closed conformation, masking recognisable epitopes, and is less likely to be stabilised in any given conformation (Munro et al., Reference Munro, Gorman, Ma, Zhou, Arthos, Burton, Koff, Courter, Smith, Kwong, Blanchard and Mothes2014). The ability to recognise HIV-1 Env conformations by smFRET therefore not only provides insights into the process of receptor binding and viral entry but also allows predictions that can inform future vaccines and anti-viral targets.
The influenza haemagglutinin protein
The influenza hemagglutinin glycoprotein is required for binding sialylated glycoproteins and glycolipids on the surface of endothelial cells via the receptor-binding domain (HA1) and the resulting clathrin-mediated endocytosis (Sieczkarski and Whittaker, Reference Sieczkarski and Whittaker2005; Eierhoff et al., Reference Eierhoff, Hrincius, Rescher, Ludwig and Ehrhardt2010) or macropinocytosis (de Vries et al., Reference de Vries, Tscherne, Wienholts, Cobos-Jiménez, Scholte, García-Sastre, Rottier and de Haan2011) of the viral particle into the cell. HA consists of three monomers of HA1 and HA2; each HA1 subunit contains a receptor-binding domain, which comes together to form the globular head of the protein, whereas the three HA2 subunits anchor the protein to the membrane and carry the fusion peptide. X-ray structures of HA (Carr and Kim, Reference Carr and Kim1993) suggest a model of fusion whereby the low pH in the endosome triggers a conformational change in HA1 which exposes the fusion peptide, followed by its extension and insertion into the target membrane (Hamilton et al., Reference Hamilton, Whittaker and Daniel2012; Liu et al., Reference Liu, Zhang, Tian, Zhao, Liu, Sun, Xiao, Zhang, Wang and Pang2012).
In order to confirm the conformational changes during membrane fusion, Das et al. (Reference das, Nikić-Spiegel, Krammer, Lemke, Munro and Munro2018) introduced donor and acceptor fluorophores to non-canonical amino acids synthesised at positions 17 in the stalk, and 127 in the fusion peptide of an H5N1 HA. The authors were able to use smFRET to distinguish irreversible conformational changes between pre-fusion and post-fusion HA2, as well as two reversible intermediate HA2 conformations. At pH 7, where HA2 is thought to be in the pre-fusion state, a high-FRET state predominated (0.92 on average), thought to represent an actual distance of 40 Å between fluorophores. The authors suggest the high-FRET state represents the pre-fusion state, where HA2 fusion peptide sits medially within the trimer (Wilson et al., Reference Wilson, Skehel and Wiley1981). By decreasing the pH, the intermediate-low-FRET state (0.53–0.2) occupancy increased, which may represent conformations sampled by HA2 during acidification of the endosome and extension of the fusion loop distal to the viral membrane, consistent with the ‘spring-loaded’ hypothesis (Carr and Kim, Reference Carr and Kim1993; Bullough et al., Reference Bullough, Hughson, Skehel and Wiley1994). However, the transitions into different FRET states remained, suggesting reversibility of the conformational changes until, at pH 5.6, the occupancy of the low-FRET state became irreversible. The authors suggest the low-FRET state represents the stable coiled-coil post-fusion HA2 induced by the acidic endosome (Bullough et al., Reference Bullough, Hughson, Skehel and Wiley1994), and the intermediate conformations serve to prevent HA2 from stabilising before the fusion target is available. By incubating the virions in liposomes displaying sialic acid receptors, the dynamics and occupancy of the low-FRET state increased, which suggests that proximity to the membrane and target receptors also increase the likelihood of the post-fusion HA2 conformation (Das et al., Reference das, Nikić-Spiegel, Krammer, Lemke, Munro and Munro2018). smFRET has therefore been useful in validating hypotheses put forward from protein structures and relating these to function.
The Ebola glycoprotein
The Ebola glycoprotein (GP) is a trimer made up of GP1 and GP2 heterodimers encompassed by a glycan cap, responsible for facilitating engagement with cells, cellular receptor binding, and promoting fusion of the viral and endosomal membranes required for virus entry (Carette et al., Reference Carette, Raaben, Wong, Herbert, Obernosterer, Mulherkar, Kuehne, Kranzusch, Griffin, Ruthel, Cin, Dye, Whelan, Chandran and Brummelkamp2011; Das et al., Reference das, Bulow, Diehl, Durham, Senjobe, Chandran, Luban and Munro2020). After cellular internalisation, Ebola is trafficked through the endocytic pathway where GP1 is cleaved to remove the mucin-like domain (muc) and glycan cap, revealing the receptor-binding site for the cellular Niemann-Pick C1 protein (NPC1) (Miller et al., Reference Miller, Obernosterer, Raaben, Herbert, Deffieu, Krishnan, Ndungo, Sandesara, Carette, Kuehne, Ruthel, Pfeffer, Dye, Whelan, Brummelkamp and Chandran2012). GP undergoes at least two major refolding processes, proceeding from pre-fusion (Zhao et al., Reference Zhao, Ren, Harlos, Jones, Zeltina, Bowden, Padilla-Parra, Fry and Stuart2016) to intermediate (Misasi et al., Reference Misasi, Gilman, Kanekiyo, Gui, Cagigi, Mulangu, Corti, Ledgerwood, Lanzavecchia, Cunningham, Muyembe-Tamfun, Baxa, Graham, Xiang, Sullivan and McLellan2016) to post-fusion conformations (Lee et al., Reference Lee, Fusco, Hessell, Oswald, Burton and Saphire2008; Pallesen et al., Reference Pallesen, Murin, de Val, Cottrell, Hastie, Turner, Fusco, Flyak, Zeitlin, Crowe, Andersen, Saphire and Ward2016), which eventually results in fusion of the host and viral membranes (Weissenhorn et al., Reference Weissenhorn, Carfí, Lee, Skehel and Wiley1998; Malashkevich et al., Reference Malashkevich, McNally, Milhollen, Pang, Kim and Kim1999). Atomic structures of pre- and post-fusion GP have been characterised; however, how conformational dynamics of GP promote Ebola virus entry is unknown.
A study by Durham et al. (Reference Durham, Govindan, Senjobe, Fels, Diehl, Luban, Chandran, Munro and Munro2020) measured smFRET between donor and acceptor fluorophores in the GP1 and GP2 N-termini, to explore the interactions between GP domains. The authors used pseudoviruses with mutant GP expressed either without the GP1 muc domain (GPΔmuc) or without the transmembrane domain of GP2 (GPΔTM). The GPΔTM structure, which cannot fuse with the cellular membrane, displayed a high occupancy of the high-FRET state. This suggests the high-FRET state is the predominant pre-membrane fusion conformation in the endosome, where GP domains are separated by 30 Å. The removal of muc and cleavage of the glycan cap increased the low-intermediate-FRET state occupancy and decreased overall transitions between states. In the presence of NCP1, the conformation transitions towards an intermediate-FRET, which may represent the final post-fusion conformation (Durham et al., Reference Durham, Govindan, Senjobe, Fels, Diehl, Luban, Chandran, Munro and Munro2020). Interestingly, the authors showed the weakly neutralising antibody KZ52 that binds proximal to the fusion loop stabilised GP in the low-FRET states, whereas ADI-15878 and ADI-15742 that contact GP1 and GP2 stabilised high-FRET (Durham et al., Reference Durham, Govindan, Senjobe, Fels, Diehl, Luban, Chandran, Munro and Munro2020). This may suggest that neutralising antibodies work to prevent the intermediate-FRET post-fusion conformation of GP.
Another smFRET study by Das et al. (Reference das, Bulow, Diehl, Durham, Senjobe, Chandran, Luban and Munro2020), where both fluorophores were placed on GP2, revealed that the glycan cap cleavage did not alter the FRET state or distance between fluorophores in GP2. The authors also labelled the N terminus of GP1 and the CX6CC domain in GP2 to take smFRET measurements during cellular membrane fusion, confirming a decreasing FRET efficiency from pre- to post-fusion, which may represent the presence of the fusion peptide to the cellular membrane. Collectively, observing conformational modulations of GP2 by endosomal pH, Ca2+, NPC1, and glycan cap removal via smFRET imaging has started to reveal roles of these factors in Ebola virus entry, and smFRET studies will continue to help fill our knowledge gap on the conformational dynamics of Ebola GP.
The SARS-CoV-2 spike protein
The spike protein (S) is a homotrimeric glycoprotein made up of three single-chain polyproteins, which protrude from the surface of SARS-CoV-2 viral particles. The polyprotein precursors are proteolytically cleaved by furin proteases into a trimer (Hoffmann et al., Reference Hoffmann, Kleine-Weber and Pöhlmann2020a, Reference Hoffmann, Kleine-Weber, Schroeder, Krüger, Herrler, Erichsen, Schiergens, Herrler, Wu, Nitsche, Müller, Drosten and Pöhlmann2020b) consisting of an S1 receptor-binding domain (RBD) and S2 membrane fusion subunit (Tortorici and Veesler, Reference Tortorici and Veesler2019). The S1 subunit binds to the cellular receptor human angiotensin-converting enzyme 2 (ACE2) (Letko et al., Reference Letko, Marzi and Munster2020), and the N terminus of S2 is proteolytically cleaved again by cellular proteases such as TMPRSS2 (Matsuyama et al., Reference Matsuyama, Nagata, Shirato, Kawase, Takeda and Taguchi2010; Hoffmann et al., Reference Hoffmann, Kleine-Weber, Schroeder, Krüger, Herrler, Erichsen, Schiergens, Herrler, Wu, Nitsche, Müller, Drosten and Pöhlmann2020b) or cathepsins B/L (Simmons et al., Reference Simmons, Gosalia, Rennekamp, Reeves, Diamond and Bates2005; Hashimoto et al., Reference Hashimoto, Sakamoto, Deguchi, Yi, Sano, Hotta, Takahashi, Yamanaka and Takayama2021), releasing the fusion peptide and enabling membrane fusion.
Cryo-EM (Cai et al., Reference Cai, Zhang, Xiao, Peng, Sterling, Walsh, Rawson, Rits-Volloch and Chen2020; Hwang et al., Reference Hwang, Lim, Yu, Kong, Sefik, Xu, Harman, Kim, Lee, Li and Flavell2020; Walls et al., Reference Walls, Park, Tortorici, Wall, McGuire and Veesler2020) has been used alongside tomography (Ke et al., Reference Ke, Ke, Oton, Qu, Cortese, Zila, McKeane, Nakane, Zivanov, Neufeldt, Cerikan, Lu, Peukes, Xiong, Kräusslich, Scheres, Bartenschlager and Briggs2020) and subtomogram averaging (Ke et al., Reference Ke, Ke, Oton, Qu, Cortese, Zila, McKeane, Nakane, Zivanov, Neufeldt, Cerikan, Lu, Peukes, Xiong, Kräusslich, Scheres, Bartenschlager and Briggs2020; Turoňová et al., Reference Turoňová, Sikora, Schürmann, Hagen, Welsch, Blanc, von Bülow, Gecht, Bagola, Hörner, van Zandbergen, Landry, de Azevedo, Mosalaganti, Schwarz, Covino, Mühlebach, Hummer, Krijnse Locker and Beck2020) to distinguish at least three different conformations of the spike protein. The trimer is thought to transition from a closed and inactive form where all three RBDs are facing downwards (Ke et al., Reference Ke, Ke, Oton, Qu, Cortese, Zila, McKeane, Nakane, Zivanov, Neufeldt, Cerikan, Lu, Peukes, Xiong, Kräusslich, Scheres, Bartenschlager and Briggs2020; Turoňová et al., Reference Turoňová, Sikora, Schürmann, Hagen, Welsch, Blanc, von Bülow, Gecht, Bagola, Hörner, van Zandbergen, Landry, de Azevedo, Mosalaganti, Schwarz, Covino, Mühlebach, Hummer, Krijnse Locker and Beck2020) to a pre-fusion conformation in which one or two of the RBDs are rotated to face upwards (Cai et al., Reference Cai, Zhang, Xiao, Peng, Sterling, Walsh, Rawson, Rits-Volloch and Chen2020; Walls et al., Reference Walls, Park, Tortorici, Wall, McGuire and Veesler2020), and finally to a post-fusion structure where all three RBDs are found facing upwards towards ACE2 (Lan et al., Reference Lan, Ge, Yu, Shan, Zhou, Fan, Zhang, Shi, Wang, Zhang and Wang2020; Shang et al., Reference Shang, Shi, Wan, Luo, Aihara, Geng, Auerbach, Li and Li2020b; Wang et al., Reference Wang, Zhang, Wu, Niu, Song, Zhang, Lu, Qiao, Hu, Yuen, Wang, Zhou, Yan and Qi2020; Yan et al., Reference Yan, Zhang, Li, Xia, Guo and Zhou2020). MD simulations further suggest a mechanism for transitions between states by predicting enhanced flexibility in the pre-fusion structure around three ‘joints’ in the stalk region, whereas the post-fusion conformation is thought to be relatively inflexible (Cai et al., Reference Cai, Zhang, Xiao, Peng, Sterling, Walsh, Rawson, Rits-Volloch and Chen2020). Adding to this, smFRET measurements on labelled SARS-CoV-2 spike proteins on the surface of recombinant lentivirus or virus-like particles showed that the spike exhibited at least four different conformations, with low (0.1), intermediate (0.3 and 0.5), and high (0.8) FRET (Lu et al., Reference Lu, Uchil, Li, Zheng, Terry, Gorman, Shi, Zhang, Zhou, Ding, Gasser, Prévost, Beaudoin-Bussières, Anand, Laumaea, Grover, Liu, Ho, Mascola, Finzi, Kwong, Blanchard and Mothes2020). The intermediate-FRET value (0.5) was most abundant and thought to represent the closed trimer with RBDs facing down, whereas a low-FRET state (0.1) was most common in the presence of ACE2 receptors, suggesting the low-FRET state represents the post-fusion conformation with all three RBDs facing up (Figure 4). The authors found that the closed conformation usually transitioned through the intermediate-FRET population (0.3) before binding to ACE2. However, the high-FRET (0.8) state did not appear to be sequential with the opening of the RBDs, suggesting that this conformation may be an off-pathway closed structure aiding immune evasion. Interestingly, convalescent plasma stabilised the spike protein in either the low-FRET (0.1) ‘open RBD’ conformation or the intermediate-FRET (0.5) ‘closed RBD’ conformation, supporting the theory that there may be an evolutionary advantage to the fourth conformation.

Figure 4. Schematic model of SARS-CoV-2 spike protein dynamics during ACE2 binding. The spike protein is a trimer consisting of an S1 receptor-binding domain (RBD) and an S2 membrane fusion subunit. S1 binds to the cellular receptor ACE2. smFRET experiments suggest a model whereby the spike transitions from a closed form where all 3 RBDs are facing downwards (FRET ~0.5) to a pre-fusion conformation in which 1 or 2 of the RBDs are rotated to face upwards (intermediate-FRET ~0.3) to a fusion structure where all 3 RBDs face upwards towards ACE2 (FRET ~0.1) (Lu et al., Reference Lu, Uchil, Li, Zheng, Terry, Gorman, Shi, Zhang, Zhou, Ding, Gasser, Prévost, Beaudoin-Bussières, Anand, Laumaea, Grover, Liu, Ho, Mascola, Finzi, Kwong, Blanchard and Mothes2020). An off-pathway high-FRET (~0.8) conformation was also observed, which was suggested to aid immune evasion (Lu et al., Reference Lu, Uchil, Li, Zheng, Terry, Gorman, Shi, Zhang, Zhou, Ding, Gasser, Prévost, Beaudoin-Bussières, Anand, Laumaea, Grover, Liu, Ho, Mascola, Finzi, Kwong, Blanchard and Mothes2020).
Emerging SARS-CoV-2 variants have been shown to have extensive mutations in the spike protein and varying binding affinities for the ACE2 receptor (Korber et al., Reference Korber, Fischer, Gnanakaran, Yoon, Theiler, Abfalterer, Hengartner, Giorgi, Bhattacharya, Foley, Hastie, Parker, Partridge, Evans, Freeman, de Silva, McDanal, Perez, Tang, Moon-Walker, Whelan, LaBranche, Saphire, Montefiori, Angyal, Brown, Carrilero, Green, Groves, Johnson, Keeley, Lindsey, Parsons, Raza, Rowland-Jones, Smith, Tucker, Wang and Wyles2020), and so could display significantly different RBD structures. A second study by Díaz-Salinas et al. (Reference Díaz-Salinas, Li, Ejemel, Yurkovetskiy, Luban, Shen, Wang and Munro2021) therefore used smFRET to compare the conformational dynamics of spike trimers from the original Wuhan-1 strain and those with the well-characterised D614G spike mutation found in the Alpha variant. While both proteins displayed a high-FRET (0.65) ‘closed RBD’ conformation and a low-FRET (0.35) ‘open RBD’ conformation in the presence of ACE2, D614G displayed fewer transitions overall and had a higher occupancy of the low-FRET open conformation, so is likely to more readily bind ACE2. Furthermore, neutralising monoclonal antibodies (mAbs) were unable to stabilise the low-FRET conformation in D614G, which could suggest greater affinity for the ACE2 receptor. A second study confirmed these results and further assessed the role of the E484K mutation originating in the Beta and Gamma variants of SARS-CoV-2 (Yang et al., Reference Yang, Han, Ding, Shi, Zhou, Finzi, Kwong, Mothes and Lu2022). In these studies, S1 was labelled with FRET pairs in D614, G614, Sα, and Sα + E484K Spike variants (Yang et al., Reference Yang, Han, Ding, Shi, Zhou, Finzi, Kwong, Mothes and Lu2022). EM and MD simulations suggest G614 and K484 reduced interactions between residues in the RBD and stabilised a disordered state more capable of binding to ACE2 (Cai et al., Reference Cai, Xiao, Lavine, Rawson, Peng, Zhu, Anand, Tong, Gautam, Lu, Sterling, Walsh, Rits-Volloch, Lu, Wesemann, Yang, Seaman, Chen and Chen2021; Gobeil et al., Reference Gobeil, Janowska, McDowell, Mansouri, Parks, Stalls, Kopp, Manne, Li, Wiehe, Saunders, Edwards, Korber, Haynes, Henderson and Acharya2021). While D614G mutants induced intermediate-FRET states (0.3–0.5), Sα and Sα + E484K appeared to predominate in low-intermediate-FRET (0.3) structures in the partially open conformation with one or two RBDs pointing up. Not only was the occupancy of the partially open and open states higher in these mutants, but the authors also show that the transition rate out of the open state was much slower than the closed state, compared to D614. The rate constants suggest that while Sα spent more time in the partially open conformation, D614G and E484K appear to induce more stable open conformations which they are less likely to transition out of. Although structurally it would seem the D614G and Sα + Ε484Κ improve ACE2 binding, the kinetic information revealed by smFRET suggests it may also be the result of dwell times in given conformations. A similar conclusion to the MD simulations is drawn and corroborates the importance of dynamic information in understanding receptor-binding affinity (Cai et al., Reference Cai, Xiao, Lavine, Rawson, Peng, Zhu, Anand, Tong, Gautam, Lu, Sterling, Walsh, Rits-Volloch, Lu, Wesemann, Yang, Seaman, Chen and Chen2021; Gobeil et al., Reference Gobeil, Janowska, McDowell, Mansouri, Parks, Stalls, Kopp, Manne, Li, Wiehe, Saunders, Edwards, Korber, Haynes, Henderson and Acharya2021; Yang et al., Reference Yang, Han, Ding, Shi, Zhou, Finzi, Kwong, Mothes and Lu2022). smFRET was able to suggest two different mechanisms whereby Sα and D614G mutants have achieved more efficient opening/binding to ACE2. The structural and functional consequences of different SARS-CoV-2 variants are essential for understanding viral entry and these dynamics in the presence of ACE2 and mAbs are important for therapeutic design.
smFRET measurements on viral helicases
Several viruses contain nucleic acid helicases that are responsible for unwinding replication substrates to facilitate the functions of the viral replication machinery. Viral helicases are essential for virus replication and are therefore potentially important drug targets. Helicases typically convert the chemical energy of ATP into mechanical movements that allow them to move on DNA or RNA substrates while unwinding them. smFRET studies of such proteins were first pioneered using bacterial systems (Ha et al., Reference Ha, Rasnik, Cheng, Babcock, Gauss, Lohman and Chu2002; Rasnik et al., Reference Rasnik, Myong, Cheng, Lohman and Ha2004), and subsequently moved to viral systems, where they have provided useful insights into the function of viral helicases during the viral lifecycle.
The hepatitis C virus NS3 protein
In the first such example of a study on a viral helicase protein, smFRET was used to study the RNA-unwinding mechanism of non-structural protein 3 (NS3), a helicase required for the replication of hepatitis C virus (Myong et al., Reference Myong, Bruno, Pyle and Ha2007) (Figure 5a). NS3 is a member of the superfamily 2 (SF2) helicases, which are able to unwind both double-strand (ds)DNA and RNA substrates. The study focused on the activity of NS3 on dsDNA since it was more tractable experimentally, while carrying a possible biological relevance due to a putative role of NS3 towards host DNA targets. Immobilised dsDNA constructs that featured a FRET pair exhibiting very close proximity and high-FRET efficiency were used; upon addition of NS3, the FRET decreased in a step-wise fashion due to the unwinding of the substrate, which led to an increase in the separation of the probes. Analysis of the kinetics of the DNA unwinding allowed the characterisation of the NS3 step sizes upon translocation and unwinding of DNA, and showed that NS3 unwinds DNA at discrete steps of three base pairs. Intriguingly, the presence of a physical translocation block (a biotin–streptavidin complex placed at the end of the processed substrate) led to a repetitive unwinding pattern, showing that part of NS3 maintains contact with the substrate, while part of NS3 translocates forward. Taken together, the results of the study indicated that the unwinding proceeded in a proposed spring-loaded mechanism where domains 1 and 2 of NS3 translocated three hidden steps consisting of one ATP hydrolysis each, while domain 3 remained fixed. This accumulated tension on the NS3-DNA complex was released by unwinding abruptly in a three base pair step (Myong et al., Reference Myong, Bruno, Pyle and Ha2007). A more recent study used smFRET combined with optical trapping to probe the translocase activity of NS3 on single-stranded DNA and RNA, and found that NS3 facilitates a novel ‘repetitive looping’ of single-stranded DNA (Figure 5b) (Lin et al., Reference Lin, Tritschler, Lee, Gu, Rice and Ha2017).

Figure 5. smFRET insights into translocation of the NS3 helicase. a) Structure of NS3 (pdb: 1CU1). The complex consists of the helicase domain (blue), a protease domain (orange), and small viral protein NS4A (cyan), which is needed for protease functionality and membrane binding. b) smFRET was used to demonstrate NS3-mediated repetitive looping on DNA. In the model, NS3 binds at the 5′ partial duplex junction and starts to translocate along the single-stranded DNA until it reaches the 5′ end where a donor dye is positioned. Once NS3 reaches the end, it snaps back to the duplex junction and re-initiates translocation (Lin et al., Reference Lin, Tritschler, Lee, Gu, Rice and Ha2017).
The vaccinia NPH-II protein
Vaccinia NPH-II belongs to the same superfamily of helicases as NS3 and unwinds both dsDNA and RNA to facilitate viral replication (Gross and Shuman, Reference Gross and Shuman1996; Raney et al., Reference Raney, Sharma, Moustafa and Cameron2010). During the initiation of unwinding, hundreds of ATPs are consumed before NPH-II progresses on the RNA template (Shuman, Reference Shuman1992; Jankowsky et al., Reference Jankowsky, Gross, Shuman and Pyle2000). Fairman-Williams et al. set out to characterise NPH-II binding to a 3′-RNA overhang that represents an in vitro unwinding substrate, as well as single and double-stranded RNAs (And et al., Reference And, Specificity and Shuman1993; Gwack et al., Reference Gwack, Kim, Han and Choe1996; Tai et al., Reference Tai, Chi, Chen and Hwang1996; Jankowsky et al., Reference Jankowsky, Gross, Shuman and Pyle2000; Fairman-Williams and Jankowsky, Reference Fairman-Williams and Jankowsky2012). Ultracentrifugation of the RNA-protein complexes in the presence or absence of ATP, ADP, and ATP cycle analogues revealed that the binding specificity towards the used RNA types changed with the ATP hydrolysis state. The kinetics of the NPH-II-RNA complex during unwinding initiation could be resolved by end-labelling of the 3′-overhang with a Cy3/Cy5 donor–acceptor pair and observing different conformation with distinct FRET efficiencies. These measurements demonstrated that the ATP hydrolysis states – rather than being associated with certain conformations – affected their relative occupancy by modulating the interconversion rates. Moreover, transient interactions were detected that could not be captured by complex ultracentrifugation under the same conditions. The authors conclude that ATP hydrolysis during unwinding initiation enables NPH-II to alternate between binding single- and double-stranded regions without losing contact with the RNA substrate and speculate that this process serves to find the conformation that allows for duplex unwinding to proceed.
smFRET insights into the HIV-1 reverse transcription machinery
Reverse transcription of the HIV-1 single-stranded (ss) RNA genome into double-stranded (ds) DNA so that it can be integrated into the host genome is an essential early step in viral replication and a major target for current anti-retroviral therapies. In the first step of reverse transcription, (−) ssDNA is partially synthesised from the viral RNA template using tRNA as a primer. In a second step, the transcribed (−) ssDNA realigns with the complementary transactivation response (cTAR) region at the 3′ end of RNA. After template re-alignment, (−) ssDNA synthesis proceeds, followed by digestion of RNA and synthesis of (+) ssDNA. Finally, the ssDNA is extended into dsDNA, representing a complete double-stranded DNA copy of the original viral RNA genome, which is then incorporated into the host’s genome by the enzyme integrase. From here, new RNA copies of the viral genome can be made via host-cell transcription. smFRET has provided important insights into many of the key proteins and processes involved in this multistep process.
The HIV-1 nucleocapsid
The HIV-1 nucleocapsid (NC) is a 55-amino-acid zinc-binding protein that facilitates the rearrangement of nucleic acid structures to thermodynamically more stable conformations (Levin et al., Reference Levin, Mitra, Mascarenhas and Musier-Forsyth2010). In the absence of NC, the viral RNA and (−) ssDNA TAR regions fold into hairpin structures that prevent intermolecular annealing (You and McHenry, Reference You and McHenry1994; Coffin et al., Reference Coffin, Hughes, Varmus, Coffin, Hughes and Varmus1997; Guo et al., Reference Guo, Henderson, Bess, Kane and Levin1997; Kim et al., Reference Kim, Nienhaus, Ha, Orr, Williamson and Chu2002). Studies employing ensemble techniques have demonstrated that NC binding shifts the TAR hairpin structures towards an open conformation, but could not resolve specific conformations due to averaging (Bernacchi et al., Reference Bernacchi, Stoylov, Piémont, Ficheux, Roques, Darlix and Mély2002; Azoulay et al., Reference Azoulay, Clamme, Darlix, Roques and Mely2003; Beltz et al., Reference Beltz, Azoulay, Bernacchi, Clamme, Ficheux, Roques, Darlix and Mély2003; Hong et al., Reference Hong, Harbron, O’Connor, Guo, Barbara, Levin and Musier-Forsyth2003).
Cosa et al. (Reference Cosa, Harbron, Zeng, Liu, O’Connor, Eta-Hosokawa, Musier-Forsyth and Barbara2004) conducted the first smFRET experiments on immobilised 64-nt TAR DNA hairpins, labelling synthetic oligonucleotides with a donor (Cy3) and an acceptor (Cy5) on each end (Figure 6a). Apart from the native TAR hairpin that forms four internal loops (L1–L4), they used mutated sequences without some of those loops. In the absence of NC, the native TAR FRET traces were static (FRET ~1), indicating a short end-to-end distance in agreement with a closed hairpin (C). Adding NC decreased the observed FRET signal, indicating an increased end-to-end distance consistent with a more open conformation (state ‘Y’). Further, the dynamics and distribution of E pointed to several conformations in the presence of NC. Only deleting the terminal loops (L1, L2) resulted in TAR in the ‘C’ form, illustrating the importance of individual loops on NC activity. Cross correlation of single-molecule donor–acceptor intensity revealed hairpin opening–closing dynamics in the millisecond range for hairpins with L1 or L1/L2 in the presence of NC. When the equilibrium was shifted towards ‘C’ by increasing MgCl2, two distinct FRET values were observed, ensuring that the aforementioned rates corresponded to the interconversion of ‘C’ and ‘Y’. These slow conformational dynamics could not be detected by previous approaches using fluorescence correlation spectroscopy (FCS) (Azoulay et al., 2003; Beltz et al., Reference Beltz, Azoulay, Bernacchi, Clamme, Ficheux, Roques, Darlix and Mély2003). However, those studies had found conformational fluctuations in the microsecond range that Cosa et al. (Reference Cosa, Harbron, Zeng, Liu, O’Connor, Eta-Hosokawa, Musier-Forsyth and Barbara2004) confirmed by donor–acceptor cross correlation of their data.

Figure 6. smFRET methodology to investigate the effect of the HIV-1 nucleocapsid (NC) on the stability, conformations, and reactivity of transactivation response (TAR) elements. a) Immobilised TAR DNA with FRET labels (Cy3 on the 5′ end; Cy5 on the 3′ end) and loop structures (L1-L4). ‘B’: Biotin label for surface attachment, ‘C’: closed conformation, ‘Y’: open conformation. The distance between the FRET labels changes upon NC addition, favouring state ‘Y’ (Cosa et al., Reference Cosa, Harbron, Zeng, Liu, O’Connor, Eta-Hosokawa, Musier-Forsyth and Barbara2004). b) Experiment on the structural effect of NC on a TAR DNA–RNA duplex (Wang et al., Reference Wang, Yeh and Barbara2009).TAR RNA and DNA anneal in the presence of NC. By washing with buffer, NC is removed from the duplex.
In a second in-depth study, Cosa et al. (Reference Cosa, Zeng, Liu, Landes, Makarov, Musier-Forsyth and Barbara2006) examined NC-mediated opening of the TAR hairpin over a broader range of conditions. By averaging smFRET traces over different time intervals and comparison with simulations, they demonstrated that the ‘C’ conformation of hairpins lacking L3 and L4 (−L3–L4) were indeed static without NC. With NC, −L2–L3–L4 opening and closing dynamics appeared to be a first-order reaction without any long-lived intermediates or multiple pathways. The equilibrium constant (K eq = k open/k close) between ‘C’ and ‘Y’ was determined by the autocorrelation amplitude (A AC) of the FRET signal and decreased with increasing MgCl2 concentration. On the contrary, analysis of –L3–L4 with NC showed evidence for complex secondary structure dynamics like long-lived intermediates and/or parallel pathways in the opening/closing process. To explain their results, the authors proposed alternative mechanistic models for opening and closing of the TAR hairpin that comprise at least two ‘Y’ forms (Y1,2) and possibly several ‘C’ forms, where C ➔ Y1 is the slow, rate-limiting transition.
In the light of complex TAR loop kinetics, Liu et al. (Reference Liu, Cosa, Landes, Zeng, Kovaleski, Mullen, Barany, Musier-Forsyth and Barbara2005) explored different pathways for the (−) ssDNA transfer in HIV-1 reverse transcription: In the ‘zipper’ pathway, the ends of the TAR hairpin initiate the interaction with TAR RNA, while in the ‘kissing’ pathway, loops L1–L4 form make the first contact. They used smFRET to monitor the NC-mediated interaction of TAR DNA with various DNA oligonucleotides analogous to parts of TAR RNA. To create analogues for the intermediates of both pathways, Liu et al. used short oligonucleotides. Adding a non-complementary oligonucleotide (12-N) shifted the ‘C’/‘Y’ equilibrium towards ‘C’, consistent with a reduction of the effective NC concentration by 12-N binding. However, an oligomer complementary to the 3′ end of TAR (12-C) caused another population at a FRET value of 0.5, which corresponded to the binding of 12-C and TAR and was stable at a timescale >250 milliseconds, providing evidence for the ‘zipper’ pathway. Liu et al. then acceptor-labelled a larger oligonucleotide (A-27-A) complementary to both the 5′ and 3′ end of donor-labelled TAR and observed the formation of a static, fully annealed product. Likewise, an oligonucleotide (A-24-B) designed to interact via the kissing mechanism also resulted into a stably annealed product with TAR. Hence both the ‘zipper’ and ‘kissing’ pathways were viable, while the zipper pathway progressed 5–7 times faster. Using shorter versions of these pathway analogues indicated that annealing can occur at any nucleation site in the TAR hairpin, despite previous observations that NC only destabilised the L1–L2 region (Cosa et al., Reference Cosa, Harbron, Zeng, Liu, O’Connor, Eta-Hosokawa, Musier-Forsyth and Barbara2004), while the kissing mechanism might be kinetically favoured by L1–L2 destabilisation.
The above publications either used TAR DNA alone or in combination with structural analogues of TAR RNA to circumvent the formation of large NC/nucleic acid aggregates when using full-length NC. In 2007, Liu et al. were able to control aggregation with a syringe pump system that allowed for rapid mixing of NC and non-immobilised donor-labelled hairpin and immediate delivery to the microscopic chamber, where an acceptor-labelled, complementary hairpin was immobilised, effectively monitoring the distance between the TAR 5′ end and the cTAR 3′ end (Liu et al., Reference Liu, Zeng, Landes, Kim, Zhu, Ma, Vo, Musier-Forsyth and Barbara2007). Over the course of annealing, immobilised TAR/cTAR converted from FRET = 0 to FRET = 1, while reaction intermediates were not detected with the time resolution of the scanning confocal microscope (>120 s). In the absence of aggregation, the distribution of the two states over time followed a single-exponential behaviour, pointing to a single transition state for annealing at low cTAR concentrations. Moreover, TAR/cTAR was not associated prior to annealing. The annealing rate decreased with the NC concentrations, consistent with NC-induced cooperative melting of TAR/cTAR. L1–L2 are sufficient for rapid annealing, while deleting L3–L4 did not affect the reaction rates, consistent with earlier data on TAR stability. At high concentrations of cTAR/TAR RNA, aggregation of the reactants occurred. Based on their observations, Liu et al. hypothesised that the rate-limiting step in TAR/TAR RNA annealing is local annealing at specific locations along the hairpin, facilitated by the NC-mediated Y conformation.
Wang et al. (Reference Wang, Yeh and Barbara2009) were interested in the structural changes NC induces in TAR dsDNA and DNA–RNA hybrids. In their experiments, they immobilised donor–acceptor end-labelled TAR DNA that resulted in FRET = 1 in a fully closed hairpin (Figure 6b). When cTAR or TAR RNA was added and the two strands annealed, the end-to-end distance measured by smFRET was 4–8 nm, suggesting a bent conformation. In contrast, when NC was washed away, FRET decreased to 0, consistent with the persistence length of dsDNA. Time-resolved FRET in the presence of NC revealed a rapid and reversible interconversion of high and low end-to-end distances and suggested cooperative binding of several NC molecules. By using NC lacking the basic N-terminal region (NTR) and denaturing the zinc fingers with EDTA, Wang et al. demonstrated that both structural features are required for DNA binding and proposed that the zinc fingers create bubbles where the bending can occur, while cooperative binding mediated by the NTR further increases the bending.
In another publication, Wang et al. (Reference Wang, Musier-Forsyth, Falk and Barbara2013) further characterised the structural changes with TAR dsDNA as a model system. Consistent with previous data, the FRET distribution exhibited two broad peaks (0.25, 0.65) in the presence of NC, while bending was not detected without NC. Fluorescence autocorrelation pointed to multiple conformations and interconversion pathways for NC-mediated bending, which was sequence-independent as demonstrated by reproducing the results with a different region of HIV DNA. To investigate the effects of DNA structure, mismatches were introduced to create bubbles in the dsDNA, which caused more structural fluctuations by increasing DNA flexibility. With NC present, the bubbled dsDNA bent even more, illustrating the importance of mismatches. To quantify the cooperativity mediated by the NC NTR, Wang et al. measured the dependence of the observed FRET signal on NC concentration and obtained a Hill coefficient of ~4, indicating high cooperativity of NC-mediated binding. With shorter TAR dsDNA, NC-mediated binding was greatly reduced, adding evidence for cooperative behaviour, as the required number of NCs apparently did not fit onto the truncated DNA.
The HIV-1 reverse transcriptase
The HIV-1 reverse transcriptase (RT) is a nucleic acid polymerase that uses RNA as a template to synthesise double-stranded DNA for integration into the host genome. RT is a heterodimer consisting of a p51 and p66 subunit, where the latter contains RNA- and DNA-dependent DNA polymerase activities and an RNase H domain (Figure 7a). Crystallographic studies of RT-nucleic acid complexes showed a single primer/template binding mode but ensemble kinetic studies suggested a heterogeneous mixture of several binding modes. In order to determine the existence of several species and obtain functional and structural information of each, early work on RT was carried out using a multi-parameter fluorescence detection technique (MFD) that measures FRET based on the reduction of donor lifetimes (Rothwell et al., Reference Rothwell, Berger, Kensch, Felekyan, Antonik, Wöhrl, Restle, Goody and Seidel2003). RT labelled with a donor fluorophore on the p66 domain and an acceptor-labelled primer were used to reveal three distinct states of RT-nucleic acid complexes. One of the states, not seen before by X-ray crystallography, did not incorporate nucleotides and was structurally different from the other two. The other two states were similar to the crystal structure but differed only by 5 Ǻ in the nucleic acid position from each other; notably, the two states underwent conformational changes upon binding a nucleotide, allowing the researchers to assign the structures as productive stages in DNA polymerisation.
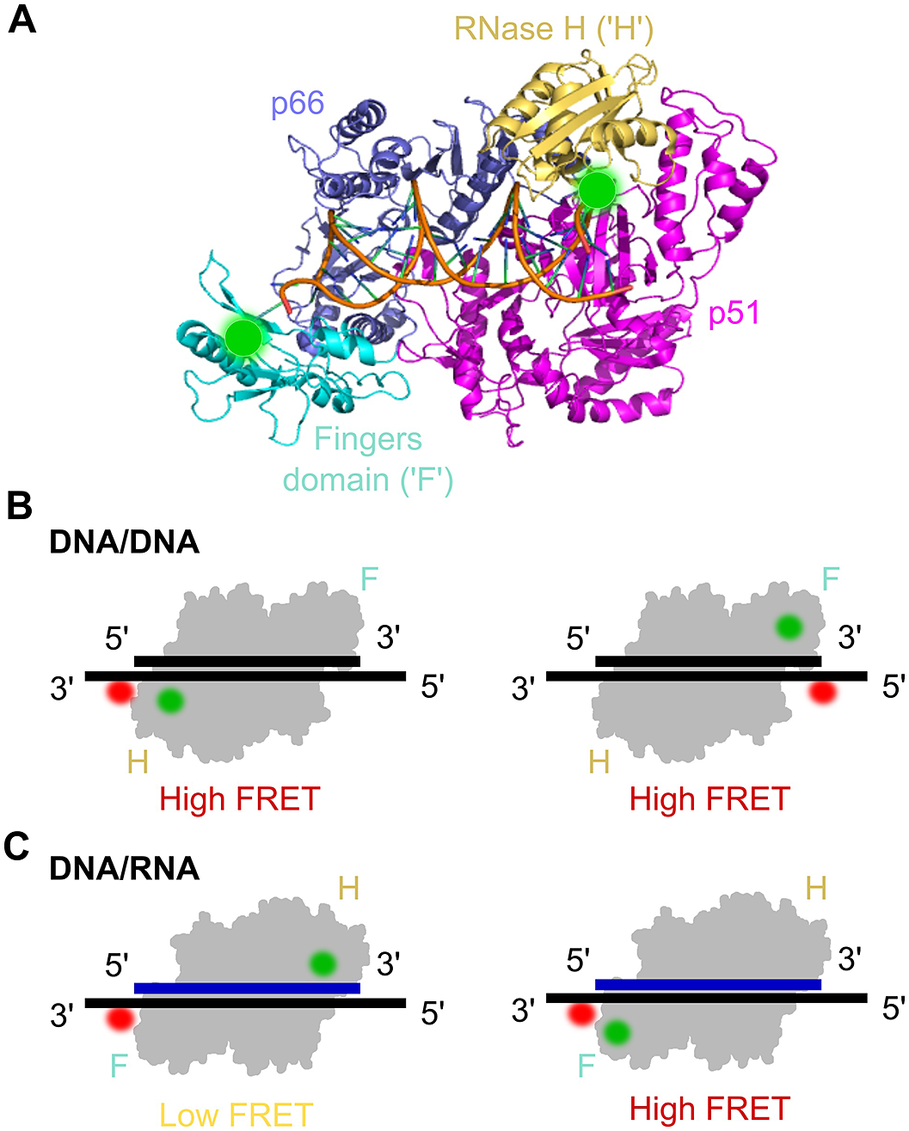
Figure 7. smFRET assay for probing the orientational dynamics of the HIV-1 reverse transcriptase (RT). a) The structure of HIV-1 RT bound to a DNA–DNA substrate (pdb: 1N6Q). RT is a dimer composed of the p66 and p51 (magenta) subunits; p66 contains the DNA polymerase (comprising the fingers (‘F’ cyan), thumb and palm domains (both blue)) and an RNase H subdomain (yellow). Labelling sites for Cy3 are highlighted by green circles. b) smFRET experiments using DNA/DNA duplex templates labelled with Cy5 were used to assess the binding orientation of RT, which suggested that RT binds DNA with the RNase H domain close to the 5′ terminus of the primer. c) Similar to panel (b) but with DNA/RNA duplexes, which suggested that the binding orientation on the RNA primer was the opposite to that on the DNA primer (Liu et al., Reference Liu, Abbondanzieri, Rausch, Le Grice and Zhuang2008, Reference Liu, Harada, Miller, Le Grice and Zhuang2010; Abbondanzieri et al., Reference Abbondanzieri, Bokinsky, Rausch, Zhang, le Grice and Zhuang2008).
RT mechanisms were also studied using smFRET in a series of three seminal papers from the lab of Xiaowei Zhuang (Abbondanzieri et al., Reference Abbondanzieri, Bokinsky, Rausch, Zhang, le Grice and Zhuang2008; Liu et al., Reference Liu, Abbondanzieri, Rausch, Le Grice and Zhuang2008, Reference Liu, Harada, Miller, Le Grice and Zhuang2010). The first paper examined the mechanism RT uses to coordinate the DNA polymerase and RNase H activities. Although RNase H cleavage analysis has shown different interaction modes with substrates, crystal structures had only shown one enzyme-binding orientation. To address this question and study RT binding conformations, a smFRET assay with millisecond ALEX was used (Abbondanzieri et al., Reference Abbondanzieri, Bokinsky, Rausch, Zhang, le Grice and Zhuang2008). Surface-immobilised DNA or RNA substrate was acceptor-labelled at the 5′- or 3′-end and immersed in a solution containing free RT molecules, donor-labelled either at the RNase H domain or at the fingers subdomain (at the polymerase domain) on the p66 subunit. The distinct FRET efficiencies showed that RT binds DNA and RNA primers in opposite directions, hence defining its enzymatic activity (Figure 7b and Figure 7c). For the binding of RT to duplexes containing polypurine RNA primers for plus-strand DNA synthesis, RT was found to dynamically switch between binding orientations. Cognate nucleotides and non-nucleoside RT inhibitors, a class of anti-HIV drugs, were found to have opposing effects on switching rates. The dynamic conformational changes of RT binding to substrates allowed the enzyme to explore multiple binding orientations, thereby regulating enzymatic kinetics and replication efficacy. This provided new insights into the dynamic structure and function of RT that has not been shown before with ensemble methods or crystal structures and could help in the development of pharmacological agents.
The second paper showed that RT can shuttle over very long lengths of double-stranded nucleic acid substrates without using any ATP (as opposed to bacterial DNA helicases such as Rep helicase that shuttle in an ATP-dependent manner), and in a thermally driven motion (Liu et al., Reference Liu, Abbondanzieri, Rausch, Le Grice and Zhuang2008). The third paper in this series focused on the transition of RT from initiation to elongation, and showed that pausing occurring during this transition is indeed due to an RNA secondary structure (Liu et al., Reference Liu, Harada, Miller, Le Grice and Zhuang2010). These fascinating contributions highlighted the surprisingly dynamic nature of proteins while interacting with nucleic acids and other substrates; the mechanistic insights and studies involving anti-viral compounds also opened avenues for drug discovery.
The HIV-1 integrase
In order to complete their infection cycle, retroviruses have to integrate the once-transcribed cDNA into the host’s chromatin. This is accomplished by the so-called pre-integration complex (PIC). Its major constituent – integrase (IN) – catalyses i) the 3′cleavage of a dinucleotide from the cDNA ends 3′ processing and ii) the insertion of cDNA into the host chromatin by strand transfer (Craigie et al., Reference Craigie, Mizuuchi, Bushman and Engelman1991; Frankel and Young, Reference Frankel and Young2003). Moreover, IN interacts with molecular import and chromatin tethering of the virus. IN functionality depends on its varying quaternary structure – dimers, tetramers, and oligomers – which is in turn influenced by the interaction with host factors like LEDGDF/p75 (lens equilibrium derived growth factor) (Van Maele and Debyser, Reference Van Maele and Debyser2005; Taltynov et al., Reference Taltynov, Desimmie, Demeulemeester, Christ and Debyser2012). Interestingly, inhibitors of LEDGDF also affect IN multimerization (Cherepanov et al., Reference Cherepanov, Maertens, Proost, Devreese, van Beeumen, Engelborghs, de Clercq and Debyser2003; McKee et al., Reference McKee, Kessl, Shkriabai, Dar, Engelman and Kvaratskhelia2008; Hendrix et al., Reference Hendrix, Gijsbers, de Rijck, Voet, Hotta, McNeely, Hofkens, Debyser and Engelborghs2011).
Borrenberghs et al. (Reference Borrenberghs, Thys, Rocha, Demeulemeester, Weydert, Dedecker, Hofkens, Debyser and Hendrix2014) set out to determine the oligomerization states in virus particles and during infection with HIV-1. To this end, they created translational fusions of IN and two fluorescent proteins (FPs) that formed a FRET pair, mTFP1 and mVenus, and incorporated them into virus particles by genetically fusing them to viral protein R (Wu et al., Reference Wu, Liu, Xiao, Kim, Seshaiah, Natsoulis, Boeke, Hahn and Kappes1995; Albanese et al., Reference Albanese, Arosio, Terreni and Cereseto2008). A series of control experiments demonstrated that IN-FP is incorporated into viruses and trans-complements catalytically active IN, while forming a heteromeric complex with it. Moreover, fluorescence co-localisation experiments with capsid protein verified that intracellular virus particles indeed contained IN-FP. By comparing the fluorescence intensity of IN-FP virus particles with purified IN-FP, the authors found that one virus particle contained about 3 to 5.5 IN-mVenus proteins and about 4–7.5 IN-mTFP1. FRET values were measured by acceptor photobleaching-mediated FRET (AP-FRET), where FRET intensity is determined by the ratio of donor intensities before and after acceptor photobleaching (rFRET). This approach to measure spatial proximity was demonstrated by incorporating the fusion protein IN-mTFP1-mVenus, which yielded an rFRET = 1.87, while IN-mTFP1 exhibited no FRET (rFRET ~1, unity). When IN-mVenus and IN-mTFP1 were co-transfected, rFRET of the resulting virus particles was 1.27, indicating the oligomerization of IN. In contrast, an IN W108G substitution adversely affecting oligomerization gave rFRET ~1, verifying that FRET increase was caused by specific IN–IN interactions. A LEDGIN inhibitor targeting the LEDGDF–IN interaction enhanced oligomerization in agreement with previous data (Kessl et al., Reference Kessl, Jena, Koh, Taskent-Sezgin, Slaughter, Feng, de Silva, Wu, le Grice, Engelman, Fuchs and Kvaratskhelia2012; Tsiang et al., Reference Tsiang, Jones, Niedziela-Majka, Kan, Lansdon, Huang, Hung, Samuel, Novikov, Xu, Mitchell, Guo, Babaoglu, Liu, Geleziunas and Sakowicz2012). IN stayed part of an oligomeric complex both in virus particles and in the host cytosol.
With the same approach (Borrenberghs et al., Reference Borrenberghs, Thys, Rocha, Demeulemeester, Weydert, Dedecker, Hofkens, Debyser and Hendrix2014), Borrenberghs et al. compared the oligomerization states of PICs in the cytosol and nucleus while using IN-eGFP and scanning confocal microscopy to quantify the amount of IN in individual PICs (Borrenberghs et al., Reference Borrenberghs, Dirix, De Wit, Rocha, Blokken, De Houwer, Gijsbers, Christ, Hofkens, Hendrix and Debyser2016). They found that in the nucleus, PICs have less IN than in the cytoplasm. However, while rFRET in cytoplasmic PICs was the same as in mature virus particles (1.27), the apparent oligomerization state in the nucleus was elevated (rFRET ~1.44). In LEDGDF/p75 knockdown cells, the oligomerization level in the nucleus was reduced to cytoplasmic levels, while the decrease of the IN/PIC ratio resembled that of WT cells. Mutating the LEDGDF/IN contact interface confirmed that the interaction with LEDGDF/p75 caused nuclear IN oligomerization, unlike the host factor TRN-SR2 potentially involved in PIC nuclear import (Christ et al., Reference Christ, Thys, de Rijck, Gijsbers, Albanese, Arosio, Emiliani, Rain, Benarous, Cereseto and Debyser2008; Taltynov et al., Reference Taltynov, Desimmie, Demeulemeester, Christ and Debyser2012). Importantly, nuclear IN oligomerization did not require LEDGDF-mediated chromatin tethering and was independent of chromosomal integration. Time-course measurements of PIC intensity in the nucleus revealed that the IN/PIC ratio was reduced upon nuclear import and subsequently remained constant. Interestingly, LEDGINs decreased the IN oligomerization state in virus particles to nuclear levels, suggesting that the LEDGIN-associated nuclear import defect may be caused by IN aggregates that prevent the PICs through the NPC. The authors proposed a model of the viral life cycle, where PICs cannot pass the NPC unless some IN is ‘shed’ – possibly as part of capsid degradation – while the IN reaching the nucleus oligomerises to form a complex capable of chromosomal integration.
Borrenberghs et al. (Reference Borrenberghs, Zurnic, de Wit, Acke, Dirix, Cereseto, Debyser and Hendrix2019) also investigated the conformation of IN oligomers in the course of a Moloney murine leukaemia virus (MLV) infection using fluorescence quantification, co-localization, and smFRET. In contrast to HIV, MLV PICs cannot enter the nucleus via NPCs. Hence, they access the host chromatin upon nuclear breakdown during mitosis (Lewis and Emerman, Reference Lewis and Emerman1994; Suzuki and Craigie, Reference Suzuki and Craigie2007; Cohen et al., Reference Cohen, Au and Panté2011). After verifying that the IN-FP fusions yielded infectious virus particles, the authors analysed the IN oligomerization inside MLV particles and found an increased rFRET (1.25), indicative of functional IN oligomerization reminiscent of HIV (Borrenberghs et al., Reference Borrenberghs, Thys, Rocha, Demeulemeester, Weydert, Dedecker, Hofkens, Debyser and Hendrix2014). The PIC numbers followed the cell cycle, with integration-dependent loss of PICs after cell division. An elevated rFRET (1.35) of nuclear PICs suggested a quaternary structure change taking place in the nucleus, which was independent of IN interaction with chromosomes or IN catalytic activity, but was instead elicited by interaction with the host factor Brd-4, a bromodomain and extraterminal domain (BET) protein (de Rijck et al., Reference de Rijck, de Kogel, Demeulemeester, Vets, el Ashkar, Malani, Bushman, Landuyt, Husson, Busschots, Gijsbers and Debyser2013; Gupta et al., Reference Gupta, Maetzig, Maertens, Sharif, Rothe, Weidner-Glunde, Galla, Schambach, Cherepanov and Schulz2013; Sharma et al., Reference Sharma, Larue, Plumb, Malani, Male, Slaughter, Kessl, Shkriabai, Coward, Aiyer, Green, Wu, Roth, Bushman and Kvaratskhelia2013). The authors conclude that the ternary complex of BET proteins, chromatin, and IN triggers a structural rearrangement within the IN complex that aides in determining the MLV integration sites.
smFRET studies of the influenza RNA-dependent RNA polymerase
Influenza transcribes and replicates its eight single-stranded, negative sense RNA segments with the viral RNA-dependent RNA polymerase (RdRp) (te Velthuis and Fodor, Reference Te Velthuis and Fodor2016). RdRp is a heterotrimeric protein complex made up of three major subunits: the polymerase itself (PB1); PB2 that contains the cap-binding domain; and PA that contains the endonuclease domain (Fodor, Reference Fodor2013). Together, the PB2 cap-binding and PA endonuclease domains cleave capped host RNAs and ‘snatch’ the 7-methylguanosine cap required as a transcription primer. The 5′ and 3′ ends of each genomic viral (v)RNA segment contain conserved nucleotides which are partially complementary to each other and bind together to form a double-stranded promoter that is bound by the RdRp (Robertson, Reference Robertson1979; Desselberger et al., Reference Desselberger, Racaniello, Zazra and Palese1980). The remaining circularised vRNA is bound along its length by nucleoprotein (NP), to form a viral ribonucleoprotein (RNP) complex (Fodor, Reference Fodor2013).
In the first smFRET study on influenza, Tomescu et al. (Reference Tomescu, Robb, Hengrung, Fodor and Kapanidis2014) purified RdRp using an insect-cell system and studied binding to vRNA terminal sequences, combining smFRET and alternating-laser excitation (ALEX). This method allowed the distances between fluorophores on the RNA to be measured during RdRp binding, which suggested that the conformational changes during RdRp binding occur in the proximal rather than the distal region of the promoter. The authors determined the molecular distances between fluorophores at different positions and used these results to ab initio model the structure of the promoter in 3D, revealing a corkscrew structure (Tomescu et al., Reference Tomescu, Robb, Hengrung, Fodor and Kapanidis2014). RdRp binding to vRNA results in either transcription of the viral genome into capped and polyadenylated mRNA or replication to form complementary cRNA, which in turn act as templates for replication (Fodor, Reference Fodor2013). During replication, the polymerase priming loop provides support for the formation of a pppApG dinucleotide formed on the terminal positions U1 and C2 of the 3′ vRNA (terminal initiation) (Ng et al., Reference Ng, Arnold and Cameron2008; Te Velthuis, Reference Te Velthuis2014). In contrast, when the intermediate cRNA becomes the template for new vRNA, the initial pppApG dinucleotide occurs on U4 and C5 (internal initiation).
Crystal structures of the RdRp show the 5′ vRNA termini bound in the active site, while the 3′ termini appears to bind to the RdRp on the surface (Reich et al., Reference Reich, Guilligay, Pflug, Malet, Berger, Crépin, Hart, Lunardi, Nanao, Ruigrok and Cusack2014), which suggests there could be multiple conformations during RdRp–promoter binding. Robb et al. (Reference Robb, te Velthuis, Wieneke, Tampé, Cordes, Fodor and Kapanidis2016) used smFRET to explore the conformations of the RdRp–promoter complex during the initiation of replication (Figure 8a). The authors showed that in the absence of RdRp, the dsRNA promoter produces a single FRET distribution centred at ~0.47, in keeping with the value expected from dsRNA. When RdRp was added, the bound conformation produced two FRET populations; a low-FRET state representing the 3′ vRNA terminus bound within the active site, and a higher FRET state representing the surface-bound 3′ vRNA. Upon addition of ApG to mimic the production of the initiation nucleotide that initiates RNA synthesis, there was an increase in the low-FRET occupancy of the RdRp–promoter complex, thought to be the active site complex which is the expected conformation for the initiation of RNA synthesis. This suggests that initial binding of RdRp with the dsRNA promoter forms a ‘pre-initiation’ conformation where 3′ RNA termini interact with the surface of RdRp before entering the polymerase active site (Figure 8b). Then, the addition of the replication initiation product stabilises the interaction with the RdRp active site and the promoter duplex unwinds.

Figure 8. Insights into influenza replication initiation from smFRET. a) Structural model of the influenza RNAP (PDB code 5MSG) bound to vRNA in a pre-initiation state where the 3′ vRNA is located on the outer surface of the RNAP, or an initiation state where the 3′ vRNA enters the RNAP active site (Robb et al., Reference Robb, te Velthuis, Wieneke, Tampé, Cordes, Fodor and Kapanidis2016). b) Model of pre-initiation, initiation, and elongation states on a vRNA template. Upon RNAP binding, the vRNA promoter exists in equilibrium between a pre-initiation state in which the 3′ RNA terminus is bound on the RNAP surface, and an initiation state in which the 3′ RNA is bound in the active site. In the presence of NTPs, the 3′ RNA starts to translocate through the active site during RNA synthesis, resulting in destabilisation of the duplex region. Insights from smFRET experiments are highlighted in red text (Robb et al., Reference Robb, te Velthuis, Wieneke, Tampé, Cordes, Fodor and Kapanidis2016). c) Model of initiation states on a cRNA template. Unlike the vRNA promoter, the cRNA promoter exhibited very limited dynamics, in part due to the longer length of the 3′ template strand of the cRNA which allows the cRNA template strand to reach further into the active site (Robb et al., Reference Robb, te Velthuis, Fodor and Kapanidis2019).
To further probe vRNA and cRNA conformations during RdRp binding, surface-bound initiation complexes were monitored before and during initiation (Robb et al., Reference Robb, te Velthuis, Fodor and Kapanidis2019). It was observed that while the 3′ terminus of the vRNA promoter is highly dynamic and transitions between pre-initiation and initiation conformations, the cRNA promoter exhibited very limited dynamics (Figure 8c). Two bases in the 3′ template strand of the cRNA promoter (absent in the vRNA promoter) allowed the cRNA template strand to reach further into the active site, limiting promoter dynamics (Robb et al., Reference Robb, te Velthuis, Fodor and Kapanidis2019). smFRET has therefore been instrumental in studying the RNA dynamics involved in influenza replication initiation, highlighting differences in the initiation mechanisms for vRNA and cRNA synthesis and providing insights into how the RNAP distinguishes these two processes.
Concluding remarks
smFRET is an important tool for deciphering conformational changes and dynamics within individual proteins and nucleic acids which would otherwise be missed by static structural characterisation, allowing for the exquisitely sensitive measurement of binding kinetics and biomolecular interactions. In this paper, we have reviewed the application of smFRET to studies of viral replication, providing an in-depth analysis of experiments carried out on a number of important pathogenic viruses, namely influenza, HIV-1, Ebola, SARS-CoV-2, hepatitis C and the vaccinia virus. Many ground-breaking experiments have been carried out to maximise the capabilities offered by this tool and have already answered multiple important questions in virus research, although a wealth of topics still remain.
Challenges to the more widespread utilisation of smFRET, such as the need for expensive hardware, specific laser-safe facilities, and specialist expertise, are being overcome by the increasing availability of more affordable open-access microscope systems (Ambrose et al., Reference Ambrose, Baxter, Cully, Willmott, Steele, Bateman, Martin-Fernandez, Cadby, Shewring, Aaldering and Craggs2020), as well as attempts within the smFRET community to publish generalised protocols and documentation to ensure the reproducibility and accuracy of smFRET measurements (Hellenkamp et al., Reference Hellenkamp, Schmid, Doroshenko, Opanasyuk, Kühnemuth, Rezaei Adariani, Ambrose, Aznauryan, Barth, Birkedal, Bowen, Chen, Cordes, Eilert, Fijen, Gebhardt, Götz, Gouridis, Gratton, Ha, Hao, Hanke, Hartmann, Hendrix, Hildebrandt, Hirschfeld, Hohlbein, Hua, Hübner, Kallis, Kapanidis, Kim, Krainer, Lamb, Lee, Lemke, Levesque, Levitus, McCann, Naredi-Rainer, Nettels, Ngo, Qiu, Robb, Röcker, Sanabria, Schlierf, Schröder, Schuler, Seidel, Streit, Thurn, Tinnefeld, Tyagi, Vandenberk, Vera, Weninger, Wünsch, Yanez-Orozco, Michaelis, Seidel, Craggs and Hugel2018; Lerner et al., Reference Lerner, Barth, Hendrix, Ambrose, Birkedal, Blanchard, Börner, Sung Chung, Cordes, Craggs, Deniz, Diao, Fei, Gonzalez, Gopich, Ha, Hanke, Haran, Hatzakis, Hohng, Hong, Hugel, Ingargiola, Joo, Kapanidis, Kim, Laurence, Lee, Lee, Lemke, Margeat, Michaelis, Michalet, Myong, Nettels, Peulen, Ploetz, Razvag, Robb, Schuler, Soleimaninejad, Tang, Vafabakhsh, Lamb, Seidel and Weiss2021). The constant development within the field will no doubt help to reduce these challenges and ensure that smFRET becomes an increasingly mainstream technique for studying biomolecular structural dynamics. This has been aided by huge improvements in cryo-EM/ET methodology in recent years, which provide structures that are essential for deciphering the dynamic and kinetic information achieved with smFRET. Further advances in computational ability, coupled with the boom in cryo-EM/ET data, have also allowed advances in molecular dynamics simulations, which will no doubt inform future dynamic structural studies (Pedebos and Khalid, Reference Pedebos and Khalid2022).
In summary, the application of smFRET to virus research has already been demonstrated to have a vast impact in furthering our understanding of how viruses replicate and interact with host systems, and provides huge potential for the design of vaccines and anti-viral treatments for existing and emerging infections.
Financial support
This research was supported by an MRC-Doctoral Training Partnership funded studentship (D.G.), a Royal Society Dorothy Hodgkin Research Fellowship DKR00620 and Research Grant for Research Fellows RGF/R1/180054 (N.C.R.), Wellcome Trust grant 110164/Z/15/Z (A.N.K.), MRC grant MR/N010744/1 (A.N.K and N.C.R) and BBSRC Grant BB/V001868/1 (C.H., N.C.R., and A.N.K.).









 Open access
Open access