


Introduction
Multiple sclerosis (MS) preferentially affects women, with clinical onset and disease course mostly occurring during the reproductive years. Although much work has been done in this area, nothing has been definitive or population-based especially with relation to current therapeutic standards for the treatment of MS. Available studiesReference Vukusic, Hutchinson and Hours1,Reference Thiel, Langer-Gould and Rockhoff2 including record linkage (e.g.Reference Lu, Dahlgren, Sadovnick, Sayao, Synnes and Tremlett3,Reference Cavalla and Gilmore4 ) on conception, during gestation and the 12 months’ postpartum are limited and plagued by limitations (e.g. sample size, inclusion criteria, teratologist input).
Historically, women with MS were often discouraged to consider pregnancy. The PRegnancy In Multiple Sclerosis (PRIMS) study was the first large study aimed at assessing the possible influence of pregnancy and delivery on the clinical course of MS.Reference Vukusic, Hutchinson and Hours1,Reference Vukusic and Confavreux5
Since the advent of formal clinical drug trials of interferon beta-1b in MS,6,7 pregnancy has been an exclusion criterion for trial entry, as with most other therapeutic trials. However, disease-modifying therapies (DMTs) are now commonly used in clinical care, raising questions about fertility and safety of DMT usage with respect to fetal exposure at conception and during gestation as well as during breastfeeding. Literature reviews have been helpful (e.g.Reference Voskuhl and Momtazee8,Reference Bove, Alwan and Friedman9,Reference Houtchens and Sadovnick10 ).
The need for a prospective population-based disease-specific pregnancy registry in MS has been discussed in detail.Reference Alwan, Chambers, Armenti and Sadovnick11,Reference Krueger, Anthony and Saltus12,Reference Dobson, Dassan, Roberts, Giovannoni, Nelson-Piercy and Brex13 To briefly summarize, a prospective pregnancy registry is an observational study specifically designed to collect information on an exposure of interest and/or disease status at pregnancy, before outcome is known. Disease-specific pregnancy registries aim to evaluate the potential for teratogenic risks associated with the disease and its associated therapies. Adopting a prospective pregnancy registry approach appears to be a very efficient method to assess clinical teratogenic potentials of neurological diseases and their medications, independently and interactively. Participants are voluntarily enrolled at conception or during early gestation until the end of pregnancy, or for a defined period afterward. Information is then collected on the outcomes and their frequency evaluated relative to an unexposed comparison group or to a valid reference population(s).14 This is in contrast to “exposure pregnancy registries” which are post-marketing surveillance studies to detect the teratogenic potential of specific marketed drugs in humans. These do exist for MS patients on specific therapies but are known to have many methodological issues.Reference Alwan, Chambers, Armenti and Sadovnick11,Reference Krueger, Anthony and Saltus12
Unfortunately, “exposure pregnancy registries” (whether retrospective or prospective) provide limited or no information on factors that can potentially separately or together result in an adverse pregnancy outcome including: maternal age; previous pregnancy history; family history; ethnicity; medications in addition to specific DMT (including “social” drugs); comorbidities (including gynecological), access to medical care for pregnant woman; methods of delivery (home, hospital, natural, C-section, etc.); and prenatal screening. In addition, DMTs available outside of “clinical trials” do not have the same level of vigilance in inclusion/exclusion criteria, that is, not as stringent as within “clinical trials” (e.g. disability, disease type, comorbidities, etc.).
Canadian Multiple Sclerosis Pregnancy Prospective Cohort Study (CANPREG-MS)
CANPREG-MS will follow pregnant women with MSReference Thompson, Banwell and Barkhof15 from any stage of conception to 1 year (12 months) postpartum, allowing existing knowledge gaps to be addressed. We anticipate that this extensive prospective registry will allow for complete assessment of real-world practices and outcomes in pregnant MS patients and, for the first time, will prospectively document pediatric outcomes in children born to MS mothers. We expect that neurologic disease outcomes may be influenced by the overall duration off DMTs, and that resumption of MS therapy postpartum may be guided by MS severity during gestation and postpartum. We anticipate that early childhood milestones may be influenced by level of maternal neurologic disability postpartum, exposure to immune-modulating medications in early pregnancy, and socioeconomic stressors.
CANPREG-MS will also follow women with MS who are contemplating a pregnancy. This will provide prospective information on fertility and factors ultimately allowing or preventing pregnancy as well as the use of assisted reproductive technologies. In classical pregnancy registries, these women are not included and yet they can provide important insight into the impact of a disease (here MS) on real-life decisions and outcomes.
Overall Objective
The overall objective is to develop a comprehensive national prospective MS pregnancy data repository (registry), encompassing a prospective patient population and to describe comparative outcomes across the patient cohorts and in children born to mothers with MS.
Outcomes
Mothers with MS
The primary maternal outcomes will be: time to conception, intrapartum and postpartum relapses, timing of discontinuation of DMTs preconception and resumption of DMTs postpartum. The secondary maternal outcomes will be: use of assisted reproduction, rate of miscarriages, medication exposure, and timing of breastfeeding in MS patients.
Liveborn Children to Mothers with MS
The pediatric outcomes will include Apgar scores at birth, developmental milestones, major/minor malformations, height/weight charts to age 1 year, illnesses/hospitalizations to age 1 year compared with babies of MS mother with no DMT exposure.
Ascertainment
Enrollment of participants comes from a variety of sources including:
1. MS neurologists and clinics affiliated with the Canadian Network of MS Clinics (CNMSC) – https://cnmsc.ca/;
2. Private MS neurologists and MS clinics not yet part of the CNMSC;
3. MS Society of Canada Research Portal (https://msresearch.ca/study/canadian-ms-pregnancy-prospective);
4. Publicity including presentations to health care professionals and MS patient groups as well as online sources (e.g. https://sadovnickresearch.med.ubc.ca);
5. Social media (Facebook, Twitter).
Multisource ascertainment is critical for relatively rare events in a given population, that is, women with MS who are pregnant or planning a pregnancy.
Complexities of Multisource Ascertainment Studies
To date (July 2019), we have 47 clinical sources referring to the study in addition to social media. Four potential sources opted out of the study for a variety of reasons (e.g. clinic director leaving Canada; retirement). Repeated attempts to contact 17 clinical sources were unsuccessful.
Obtaining input and support of neurologists and lay groups across Canada was relatively easy. However, once all the University of British Columbia (UBC) ethics hurdles were successfully completed, we did not expect major obstacles in enrolling other sites to advertise the study since all consenting, data collection, analyses, etc. would be done through UBC. All that was being asked was to display the information about the study in clinic areas. Interested persons would then directly contact UBC for information, consenting, etc.
Once the UBC ethics were fully approved, personal emails were sent by the PI to the major neurology practices/clinics across Canada. The majority of these neurologists were already aware of the study plan through direct communications at meetings of the CNMSC. Difficulties occurred:
Email replies took a long time and many follow-ups are needed;
Some sites/private neurologists did not require local ethics approval to advertise the study;
Many sites/private neurologists were uncertain about the need for local ethic approval and resolution of this took considerable time;
Some sites/private neurologists were told to submit “expedited” local ethics (at least two of these took 4 months from submission to approval);
Some sites/private neurologists had to submit full ethics reviews for their “home” base (hospital, university). These were full submissions for full board reviews. UBC personnel assisted with these as much as possible, but time from submission to approval was lengthly.
See Table 1 for a summary of these processes.
Table 1: Status of ascertaining sources active to announce study (as of July, 2019)
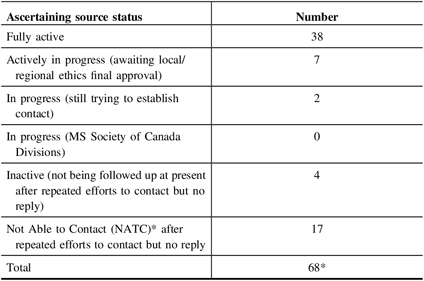
* These numbers do not include recruitment sources from public advertising, patient presentations, or social media.
Canpreg-MS Registry: Inclusion Criteria
1. Women with a confirmed diagnosis of MSReference Thompson, Banwell and Barkhof15 aged 18 years and older (19 years and older in New Brunswick);
2. Women with a confirmed diagnosis of MS who are actively planning to become pregnant OR women with a confirmed diagnosis of MS who are currently pregnant (regardless of trimester or whether or not the pregnancy was planned).
3. Canadian residents during the study period.
Consenting
MS women who have learned about CANPREG-MS can choose to contact UBC by calling a dedicated 1-800 number or by sending an email to a dedicated address. The study coordinator will then contact them to explain the study in detail and address any questions or concerns they might have. They will then be invited to participate and if agreeable, two copies of the consent form and a clinical “Release of Information” (ROI) for their physician(s) will be mailed to them.
Participants are asked to return the signed consent form and ROI in a self-addressed and stamped envelope to UBC and keep a copy for their own records. Physicians identified on the ROI (or the appropriate Regional Health Authority in Newfoundland (NL)) will then be contacted and asked to provide their medical history information. (An additional ROI will be signed after a live birth for the infant’s physician.) Data collection can only begin once the signed consents are returned to UBC.
UBC does not allow consenting by fax or email.
Data Collection at Enrollment: “CANPREG-MS Initial Intake Interview” Form
Baseline information for consented participants on demographics (e.g. date of birth, ethnicity, education), personal medical/surgical history, family history of MS and other diseases, medication history, hospital admission information, diet, reproductive history, current pregnancy (for pregnant participants), and MS history will be collected. UBC research personnel will conduct telephone interviews using standardized questionnaires.
Data Collection at Follow-up
“CANPREG-MS Follow-up Interview”
After the initial intake interview takes place, participants will be followed longitudinally. Data will be collected through telephone interviews using standardized follow-up questionnaires. A secure, web-based application REDCapReference Harris, Taylor, Thielke, Payne, Gonzalez and Conde16 (Research Electronic Data Capture) designed exclusively to support data capture for research studies has been used for developing data entry forms. The REDCap electronic capture tools are hosted and supported by UBC Advanced Research Computing.
Pregnant women will be contacted at set times during the 12 months’ postpartum and information will also be collected for all live births at set intervals for the first 12 months. See Table 2 for the data collection timelines.
Table 2: CANPREG-MS data collection schedule on pregnant women with MS
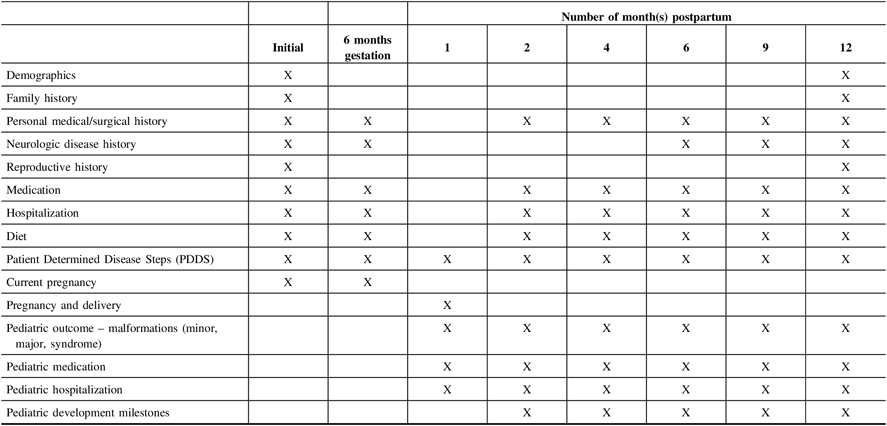
Participants planning a pregnancy will be recontacted every 6 months. They are, however, encouraged to contact the study center if any changes occur in the interim (e.g. pregnancy achieved, decision to stop trying because of MS or other reasons, etc.).
“CANPREG-MS Follow-up After Delivery”
As seen in Table 2, maternal and pediatric questionnaires will be administered at specific time points outlined. The study personnel will contact patients directly and confirmation will be obtained from the baby’s physician as appropriate based on the developmental screen and the identification of minor/major malformations. Participants will be given the option to decline being recontacted for follow-up health information.
As this study is anticipated to be comparable to the New England MS Pregnancy Registry,17 which is a regional and not national-wide registry, we will use the developmental scale designed for that study, largely adapted from the Denver Developmental Screening TestReference Frankenburg and Dodds18,Reference Frankenburg, Dodds, Archer, Shapiro and Bresnick19 commonly used in developmental assessments.
Based on the 2013 report by the Public Health Agency of Canada,Reference Irvine, Luo and Leon20 approximately 1 in 25 Canadian babies is diagnosed with 1 or more congenital anomalies every year. In 2009, the Canadian congenital anomalies prevalence rate was about 385 per 10,000 total births. We will compare rate of congenital anomalies among live births with the national rate.
Minor and major abnormalities will be identified in live births. Major malformations are defined as “structural abnormality with surgical, medical or cosmetic importance”.Reference Holmes21 Minor malformations are defined as “defects in structure with no surgical, medical or cosmetic importance”.Reference Holmes21 If a malformation (major or minor) is identified within the study period, the data will be reviewed by the two teratologists and receive a final coding as minor, major, or suggestive of a syndrome in accordance with the International Classification of Diseases or “ICD-10”22 and the Center for Disease Control Classification system.23
Re-Enrollment
As this is a 5-year study, it is possible that participants may have more than one pregnancy (or planning pregnancies) during the study period. Such women will be reconsented and entered into the study as a new participant. Although the pregnancies or attempts will be linked, essentially they will be treated as separate events.
Data and Safety Monitoring
Each participant and live birth will be assigned a code number so that his/her identity (i.e. name or any other identifying information) will stay confidential. All data entered into the study database will be de-identified. The research database will be password-protected. Processing and analysis of the de-identified data will be conducted by the PI and her authorized study personnel.
Information from the phone encounters with participants and from the clinical records will be stored in the study database at the UBC site. Records will be kept in a locked file with access limited to the PI and the authorized study personnel.
Canpreg-MS Registry: Statistical Analysis
Analyses will compare treated (exposed to DMT at conception) versus untreated (not exposed to DMT at conception) participants on the outcome measures. Categorical variables will be reported as frequencies and percentages, with mean and standard deviation reported for continuous variables. Categorical data will be compared by the χ2 test, Fisher’s exact test, or logistic regression as appropriate. Continuous data will be compared by t test or linear regression as appropriate.
At the end of year 5, the primary analyses will compare the treated versus untreated participants on each of the outcome measures, using repeated measures analysis of variance. This will be done separately for each treatment (specific therapy) to evaluate whether medication exposure has any significant impact. The ordinal and linear regression models will also be used where necessary, to incorporate other potential associated factors such as clinical MS severity, age, and disease duration, both in order to avoid confounding effects and to investigate other potential risk factors of obstetrical and pediatric outcomes.
We will not include participants who opt to adopt a child(ren) or use gestational carriers.
For analyses including DMT usage, as most are only approved for those with relapsing/remitting MS, women with other MS disease types (e.g. secondary progressive, primary progressive, relapsing progressive) will be excluded from this specific outcome.
In assessing DMT resumption, women who are planning another pregnancy (or are pregnant again) or still breastfeeding at 12 months postpartum will be excluded from this analysis if they have opted to not resume DMT for these specific reasons. The adherence of DMTs (Yes/No) will be treated as a binary outcome variable. Pearson’s χ2 test and Fisher’s exact tests will be used for comparison to explore potential risk factors such as planning another pregnancy. To access the joint effect of risk factors, we will apply logistic regression analysis. The 12-month postpartum will be the study endpoint. We will do sub-analyses throughout the study period at the various points of contact (~6 months gestation, 1, 4, and 6 months postpartum).
Secondary analysis on participants will include employment as another binary outcome variable of interest. If employed prior to pregnancy, participants’ employment status will be assessed (“Has your return to employment met your pre-pregnancy expectations for your return to employment? (Yes/No)” and if relevant, “why not” (e.g. MS, childcare issues, etc.). The 12-month postpartum will be the study endpoint.
Canpreg-MS – Other Considerations
Risk and Discomforts
The study will collect clinical, obstetrical, and pediatric information from physicians (or the appropriate Regional Health Authority in NL) for whom we have signed ROIs. We do not anticipate any risks or discomfort to CANPREG-MS participants or the babies. Only aggregated de-identified information will be analyzed.
Potential Benefits
Through CANPREG-MS, we will learn:
How often do women with MS have issues with conception?
Are there “MS-related” problems during pregnancy?
What are pregnancy outcomes taking into account factors such as MS type, MS course, DMT exposure, duration, etc.?
We will use this information to develop a patient educational resource that will be responsive to concerns of women with MS and will help them in making the personal decision about pregnancy. There are no specific benefits for the individual participants. However, information obtained will benefit our understanding of pregnant and pediatric outcomes in this population, thus benefitting the MS patients and medical communities as a whole.
Withdrawal
In accordance with consents, participation in this research study is voluntary.
Potential Pitfalls
As with all studies, potential pitfalls do exist. Participant dropout is always a concern in study design. In our clinical experience, the choices that women must make postpartum regarding their MS care (DMT resumption, breastfeeding/weaning, etc.) require clinical follow-ups, and therefore the study period would represent a time when women are more likely to follow-up for clinical and research purposes. Thus, we anticipate MS clinical visits by them during the 12 months’ postpartum and information will be available through the signed ROIs.
A higher proportion of women with MS may be planning another pregnancy, and hence there may be lower rates than expected of DMT resumption (primary outcome) among participants. There may also be other reasons to be identified for participants not resuming DMTs.
Acknowledgments
The authors gratefully acknowledge UBC study personnel – Irene Yee, Maria Criscuoli, and Kevin Atkins.
Drs. Jan Friedman and Christina Chambers are the teratologists for this study.
We appreciate the collaboration of Canadian MS Neurologists, the Canadian Network of MS Clinics, and the MS Society of Canada for assistance with recruitment.
The study would not have been possible without the volunteer participants and their efforts are sincerely acknowledged.
Funding
This study was funded by an unrestricted grant from Biogen MA Inc. (CAN-MSG-17-11120).
Disclosures
ADS has received funding from Biogen, Novartis, Genzyme Sanofi, and Teva; has received travel funds from Genzyme Sanofi and Biogen; and is on the Neurodiem Editorial Board.
RC is the Site Investigator for studies funded by Roche, Novartis, MedImmune, EMD Serono; receives research support from Teva Innovation Canada, Roche Canada, and Vancouver Coastal Health Research Institute; and has received honoraria from Roche, EMD Serono, Sanofi, Biogen, Novartis, and Teva.
MH served as a consultant for Sanofi Genzyme, Biogen, Teva, Genentech, and Serono; has unrestricted research support, as an independent investigator, from Sanofi Genzyme, Biogen, and Serono; her work is also supported by CMSC and NMSS.
AS has received honoraria from Teva, Biogen, and Sanofi Genzyme.
PS has received grant support from CIHR, Arnold P. Gold Foundation, University of Alberta Teaching and Learning Enhancement Fund, Biogen Pharmaceuticals, MS Society of Canada; has received speakers Bureau/Honoraria from the MS Society of Canada; and is on the Consulting/Ad Board for Novartis Pharmaceuticals and the Canadian Pharmacists’ Association.
Statement of Authorship
ADS was Study Principal Investigator. RC, MH, AS, and PS were Study Collaborators. ADS wrote the first draft. All authors reviewed and provided input toward the final version, prepared by ADS.


