



Introduction
Epilepsy is a chronic brain disease that is characterized by an enduring predisposition to recurrent, unprovoked epileptic seizures.Reference Fisher, Acevedo and Arzimanoglou 1 Epilepsy is the second most common neurological condition, with approximately 75,000 adults and 15,000 children (0–17 years) living with epilepsy in Ontario.Reference Bowen, Snead, Chandra, Blackhouse and Goeree 2 , 3 The Ontario Brain Institute estimates that there are approximately 6100 new physician-diagnosed cases in Ontario each year.Reference Ng 4
The first epilepsy gene, CHRNA4, was discovered in 1994. Since then, advances in molecular genetics have led to the identification of novel genes for monogenic and complex genetic epilepsies. A recent review reported nearly 1000 genes associated with epilepsy.Reference Wang, Lin and Liu 5 Genetic testing now plays a pivotal role in the clinical management of patients with epilepsy. The exact proportion of epilepsies occurring secondary to monogenic and polygenic mutations is currently unknown. However, in certain epileptic conditions, the likelihood of a genetic etiology may be higher, particularly the epileptic encephalopathies.Reference Thomas and Berkovic 6 Hence, 30% of epilepsies are drug refractory and may warrant early diagnosis through genetic testing. Thus, approximately 1830 patients per year could possibly proceed to genetic testing in Ontario, although a number of these patients might have other non-genetic demonstrable causes.
The impact of a genetic diagnosis on an individual with epilepsy can be quite significant. Accurate genetic diagnosis may guide treatments such as disease-modifying therapies and/or medications known to be effective in certain epilepsy syndromes. Genetic diagnosis may also help to prognosticate and limit further investigations that have associated risks and cost. Further, genetic diagnoses may help identify or anticipate potential co‐morbidities, allowing for optimization of treatment. With appropriate genetic diagnosis, genetic counseling for future pregnancies may be possible. Finally, a genetic diagnosis brings closure and peace of mind to the families of those with a genetic disease whether treatable or not. A negative genetic result in the diagnostic evaluation of epilepsy is equally significant. A negative test may allow the patient to proceed without delay toward critical therapies such as epilepsy surgery that would not be indicated in most genetic epilepsies. A negative genetic result, therefore, may avoid long delays to appropriate treatments.
However, potential concerns about genetic testing include psychological distress, social stigma and problems with health and life insurance. Although a significant and growing body of literature implicates specific genes and mutations in particular epileptic conditions, there is little general guidance available on the circumstances in which genetic testing is indicated and test selection in order to guide optimal test appropriateness and benefit.Reference El Achkar, Olson, Poduri and Pearl 7
The following is an account of the development of guidelines for genetic testing in epilepsy, which have been developed in Ontario, Canada, to aid clinicians in determining the indications for genetic testing (focused gene panels or comprehensive epilepsy gene panels) in adults and children with epilepsy.
Establishment of the Genetic Testing Advisory Committee (GTAC) in Ontario
Ontario (Canada) has a single-payer system of health care, which is funded by the Ministry of Health and Long-Term Care (MOHLTC) through the Ontario Health Insurance Program (OHIP). Currently, the primary means of genetic testing in epilepsy in Ontario is by targeted single-gene testing, selective gene panels or comprehensive epilepsy gene panels. At present, this epilepsy gene panel testing is funded in Ontario by OHIP for eligible patients through the MOHLTC’s out-of-country/out-of-province (OOC/OOP) prior approval program. Over the past three fiscal years, from 2014–15 to 2016–17, approximately 180 epilepsy gene panel tests were approved through this process and sent for testing OOC—usually to the US or Germany—each year at an average annual cost of approximately $1 million Canadian dollars or about $5700 Canadian dollars per test.
The process is to obtain and bank the patient’s DNA with appropriate consents and pre-testing genetic counseling and then request approval from the Ministry for Funding before the testing can be done. For whole-exome sequencing (WES), the epilepsy patient must be referred to a geneticist who is the only physician in Ontario who can order this study, which also is done out of Canada. The number of genetic tests ordered in this manner over the past 5 years in Ontario is shown in Figure 1.
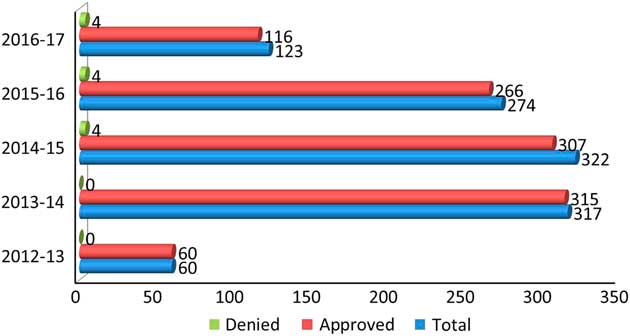
Figure 1 Number of gene panels ordered in the past 5 years in Ontario.
The GTAC was established in Ontario in April 2014 with a 3-year mandate to review the clinical utility and validity of genetic tests and provide advice to the Ontario Ministry of Health and Long-Term Care (“Ministry”) on the provision of genetic testing in Ontario. These recommendations were to be formulated in order to guide the ministry’s decision-making on public funding of genetic testing in Ontario. As part of their mandate to examine the clinical validity and utility of genetic testing in a number of disease processes, GTAC appointed a working group to develop recommendations and guidelines for genetic testing in epilepsy in particular. This working group comprised three pediatric epileptologists (OCS, ED and ANP), one adult epileptologist (DA) and four medical geneticists (DD, SG, KB and ML) from Ontario.
Requirements to Proceed to Focused Gene Panels or Comprehensive Epilepsy Gene Panels
The working group felt that it was axiomatic that the critical step in the determination of criteria for genetic testing in epilepsy is a definition of the epilepsy phenotype at issue. This includes a detailed clinical evaluation with careful documentation and delineation of seizure semiology, epilepsy syndrome, associated neurological co-morbidities, systemic manifestations, clinical course and any significant family history. This epilepsy phenotyping not only helps in selection of an appropriate genetic test but also in the interpretation of results and subsequent counseling, and further testing, if indicated.
Further investigations require a close liaison among physicians, neurologist/epileptologist, clinical geneticist and biochemical geneticist. A brief overview of this process is depicted in Figure 2. In view of these caveats, the recommendations of the GTAC working group on epilepsy for genetic testing in epilepsy were as follows.
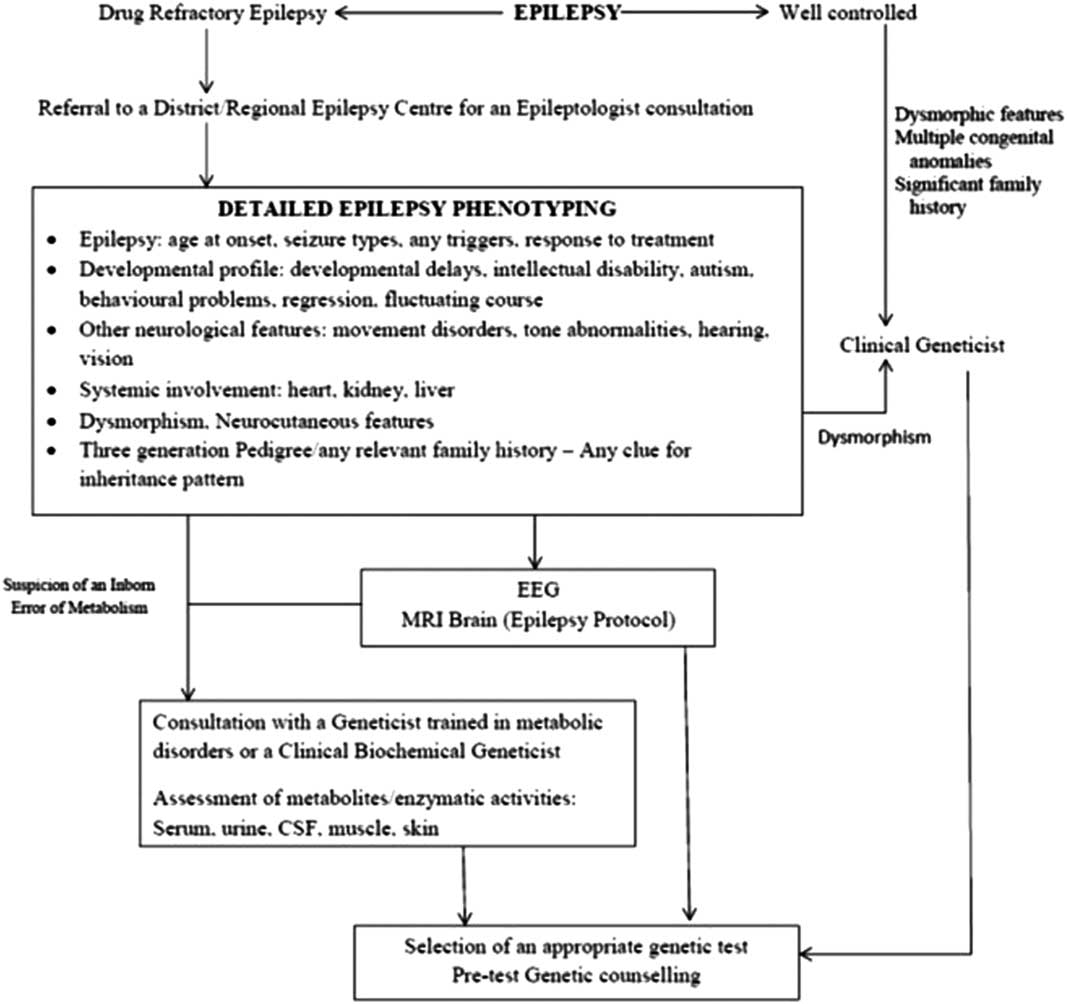
Figure 2 Flowchart depicting the prerequisites to genetic testing in patients with epilepsy.
Mandatory Prerequisites
∙ If the epilepsy is uncontrolled, referral to, or consultation with, a District or Regional Epilepsy Centre for an epileptology consultation (consistent with Provincial Guidelines) 8 is required. An epileptologist refers to an adult or pediatric neurologist with at least 6 months of training in epilepsy during his/her residency. The Ontario Epilepsy Network entails regionalization of epilepsy care into District Epilepsy Centers and Regional Epilepsy Surgery Centers of Excellence and represents a model of comprehensive epilepsy care in Ontario [population ~ 14 million] developed recently by MOHLTC. 8
∙ If epilepsy is well controlled or if there are dysmorphic features, referral to a clinical geneticist is required.
∙ If there is developmental regression or other clinical features suggestive of an inborn error of metabolism, consultation with a biochemical geneticist or a physician with training in inherited metabolic disorders is strongly recommended.
Diagnostic Procedures that are Always Indicated Before Proceeding to Focused Gene Panels, Comprehensive Epilepsy Gene Panels
∙ EEG, preferably video-EEG
∙ MRI of the brain with/without proton MR spectroscopy
∙ Consult with an epileptologist
Diagnostic Procedures that are Dependent upon Clinical Circumstance, but Not Required for Progression to focused Gene Panels or Comprehensive Epilepsy Gene Panels
∙ Metabolic: These investigations should be tailored to the specific clinical presentation (consider consultation); the most commonly used tests may include MR spectroscopy, lactate, blood gas and electrolytes, glucose, ammonia, plasma and/or CSF amino acids, total homocysteine, urine organic acids, acylcarnitine profile, sulfite screen, urine S‐sulfocysteine, uric acid, biotinidase and/or urine α‐aminoadipic semialdehyde, as well as other tests as clinically indicated.
∙ Neonatal/infantile seizures: Consider a trial of pyridoxine (B6), pyridoxal phosphate and folinic acid.
∙ CSF studies: routine cytology, glucose, lactate and amino acids; consider neurotransmitter assays in refractory epilepsy.
∙ Chromosomal microarray in the presence of developmental delay/intellectual disabilities and/or dysmorphic features.
Any of these investigations may direct the physician to consider a single-gene test or a specific gene panel—for example, specific inborn error of metabolism gene, cortical malformation gene panel or mitochondrial gene panel. Further, if any of these tests are under consideration, or if the results will require additional expertise for interpretation, a consultation with a clinical and/or biochemical geneticist may be indicated.
Biochemical Testing
Inborn errors of metabolism are relatively less frequent causes of epilepsy.Reference Mercimek-Mahmutoglu, Patel and Cordeiro 9 Seizures may dominate the clinical phenotype or be a part of the broader neurological phenotype, which usually includes developmental delays/regression, intellectual disability and/or movement disorders. Early-onset epilepsy, history of regression, multiple neurological co-morbidities, altered biochemical parameters, history of parental consanguinity and suggestive neuroimaging are few of the clues for the presence of an inborn error of metabolism. If there is such a suspicion, evaluation by a biochemical geneticist or geneticist trained in metabolic disorders is warranted. Epilepsy and inborn errors of metabolism have been reviewed recently.Reference Sharma and Prasad 10 The biochemical diagnosis is usually confirmed by genetic testing.
Who Should be Tested?
If a genetic etiology of epilepsy is suspected on the basis of epilepsy phenotyping, the patient should be referred to a District or a Regional Epilepsy Centre for expert consultations. Treatable causes should be kept in mind first and foremost. Genetic testing should not proceed in the absence of the ability to provide pre- and post-test genetic counseling. The following are indications for genetic testing in epilepsy:
∙ When the clinical features (age of onset, seizure semiology and EEG features) are consistent with a distinct epilepsy syndrome as defined by the International League Against Epilepsy (ILAE), with the exception of syndromes outlined in the following section. 11
∙ When the prognosis based on clinical and EEG findings is poor or the likelihood of lethal outcome is high.
∙ When epileptic seizures are refractory to medical treatment as defined by the ILAEReference Kwan, Arzimanoglou and Berg 12 (with no apparent acquired cause).
∙ When epilepsy is associated with features suggestive of inborn errors of metabolism.
∙ When epilepsy is associated with distinctive patterns of malformations of cortical development identified on neuroimaging studies.
∙ When epilepsy is associated with clinical signs of neurodegeneration.
∙ When epilepsy is associated with paroxysmal neurological features such as paroxysmal dyskinesias, episodic ataxias and hemiplegic migraine.
∙ When epilepsy is associated with additional syndromic features such as developmental delay/intellectual disability, multiple congenital anomalies and dysmorphic features.
∙ When familial epilepsy is present, defined as at least two first‐degree family members with related epilepsy syndromes, unless the epilepsy syndrome is benign.
Who Should Not Be Tested?
In general, genetic testing is not recommended in drug-responsive epilepsy or at epilepsy onset, although the evidence may emerge in future that early testing may be more cost-effective.Reference Ream and Patel 13 Additional specific circumstances that do not warrant genetic testing are listed below:
∙ Recognizable seizure syndrome with benign course
∙ Childhood epilepsy with centro-temporal spikes (previously termed benign rolandic epilepsy)
∙ Isolated mesial temporal lobe epilepsy with hippocampal sclerosis
∙ Typical childhood Absence epilepsy (although if it is early-onset or medically refractory epilepsy, one should consider and test for GLUT1 deficiency)
∙ Juvenile myoclonic epilepsy, which is well controlled on medications and without intellectual disability or any signs of neurodegeneration
∙ Acquired epilepsy
Which Test?
Epilepsy phenotype, neuroimaging and initial biochemical/metabolic screening may guide the selection of an appropriate genetic test. The limitations of various modalities must be considered while making this decision. Consultation with a clinical geneticist is often helpful. Commonly used testing modalities and their utility are summarized in Table 1.
Table 1 Common genetic tests used to investigate patients with epilepsy
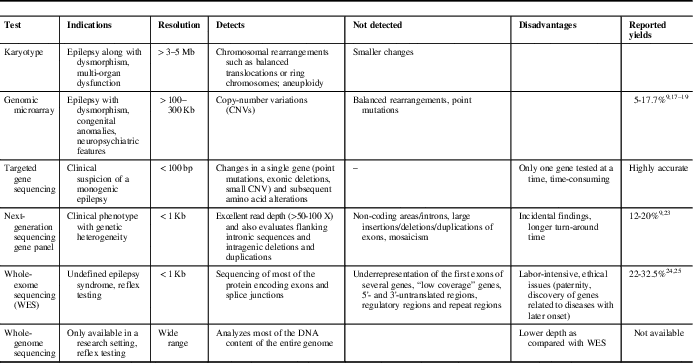
Genomic Microarray is particularly useful in patients with associated dysmorphism, congenital anomalies or neuropsychiatric features. It detects larger copy-number variations (CNVs) (>100-300 Kb). Rare CNVs represent variations in genomic DNA sequence of at least 1 Kb in length seen in <1% of the population. They are reported to account for 10% of childhood epilepsies and up to 5% of epileptic encephalopathies. 14 Most of these regions may not contain any known epilepsy genes. For example, 15q13.3 microdeletions and 16p13.11 deletions are frequently observed in patients with genetic generalized epilepsies.Reference de Kovel, Trucks and Helbig 15 Sometimes, these regions may harbor known genes (e.g., cases of neonatal seizures caused by 2q24.3 region microduplication, which harbor SCN1A, SCN2A and SCN3A genes).Reference Heron, Scheffer and Grinton 16 Further, they may cause both monogenic disease or act as risk alleles. They cannot be detected by routine karyotyping. However, microarray may miss small exonic deletions and duplications.
The reported yield of genomic microarray in the genetic evaluation of epilepsy is variable. In 805 children with epilepsy/seizures, 40 cases (5%) had their epilepsy phenotype explained by pathogenic CNVs.Reference Olson, Shen and Avallone 17 In children with drug refractory epilepsy (n=110), pathogenic deletions were reported in 13% cases by array comparative genomic hybridization.Reference Mercimek-Mahmutoglu, Patel and Cordeiro 9 In older patients with epilepsy (n=279), 26 (9.3%) had rare CNVs.Reference Striano, Coppola and Paravidino 18 In another study, the yield was reported to be 17.7% in patients (aged <23 years) with epilepsy.Reference Hrabik, Standridge and Greiner 19
Karyotype remains helpful even today to identify chromosomal rearrangements such as balanced translocations or ring chromosomes, and aneuploidy. This test should be reserved for patients with recognizable chromosomal syndromes, a family history of chromosomal rearrangement or a history of multiple miscarriages.Reference Miller, Adam and Aradhya 20
Fluorescence in situ hybridization analyzes specific chromosome portions and is usually reserved for the confirmation of microdeletion syndromes/duplication identified by other techniques (e.g., inv-dup 15).
Multiplex ligation-dependent probe amplification (MLPA) is used to identify intragenic deletions or duplications, such as sometimes observed in some patients with SCN1A- or CDKL5-related epilepsy who are negative on regular sequencing or microarray.Reference Scheffer 21 , Reference Ottman, Hirose and Jain 22
Single-gene sequencing detects changes in a single gene (point mutations, exonic deletions, small CNV) and subsequent amino acid alterations. It may be used if a classical phenotype is encountered—for example, suspected GLUT1 deficiency (SLC2A1 gene) or Dravet syndrome. It is highly accurate, but targets one gene at a time and is time-consuming. Its use becomes limited in situations where there is genetic heterogeneity and phenotypic variability. Dravet syndrome due to the SCN1A mutation is an excellent example to display these complexities involved in the genetic testing (Table 2).Reference Scheffer 21 , Reference Ottman, Hirose and Jain 22
Table 2 Dravet syndrome (DS) as an example of complexities involved in genetic testing

Next-generation sequencing (NGS) analyzes a large number of DNA segments (usually exons) of a selected group of genes organized in panels. It usually has excellent read depth (>50-100×) and also evaluates flanking intronic sequences and intragenic deletions and duplications. It is currently the most cost-effective method of detecting mutations in coding regions. However, small deletions and CNVs need to be further investigated by deletion/duplication testing or MLPA.
A total of 329 patients with early-onset drug-refractory epilepsy were screened using 30 gene- and 95 gene-panels. The overall yield was 20.3%, with a 95-gene panel giving an additional yield of 6.3%.Reference Parrini, Marini and Mei 23 Mercimek-mahmutoglu et alReference Mercimek-Mahmutoglu, Patel and Cordeiro 9 reported a yield of 12.7% in children with drug-refractory epilepsy and global developmental delay.
Whole-exome sequencing involves sequencing of most of the protein encoding exons and splice junctions. However, underrepresentation of the first exons of many genes and low coverage of some genes leads to almost 5-10% of the coding sequence in the human genome being not screened/captured by WES. Further, 5'- and 3'-untranslated regions, regulatory regions and repeat regions are poorly covered. It is to be noted that the possible NGS/WES results may require confirmation—for example, by Sanger sequencing.
In 50 patients with unexplained early-onset epileptic encephalopathy, WES showed disease-causing variants in 11 (22%) patients.Reference Allen, Conroy and Shahwan 24 The yield was up to 32.5% in another cohort of patients with undiagnosed intellectual disability with/without epileptic encephalopathy.Reference Thevenon, Duffourd and Masurel-Paulet 25
Berg et alReference Berg, Coryell and Saneto 26 explored the role of genetic testing in children with newly diagnosed epilepsy. The overall yield was 40.4% for various genetic tests performed. The yield was greater for epilepsy panels (29.2%) and WES (27.8%) than for the microarray (7.9%). Sequential testing (genomic microarray followed by epilepsy gene panel) was shown to be preferable in children with West syndrome.Reference Wirrell, Shellhaas and Joshi 27
Whole-genome sequencing analyzes most of the DNA content of the entire genome with even coverage but has lower depth than WES. It is, however, not available clinically. However, abnormalities such as triplet repeats (e.g., Fragile X) require polymerase chain reaction-based analysis, and abnormal methylation (e.g., Angelman syndrome) requires special genetic testing.
Simultaneous or sequential testing of the parents of the affected patient (trio analysis) is desirable for proper interpretation of sequencing results. However, this may be limited by the costs and insurance coverage.Reference Schwarze, Buchanan, Taylor and Wordsworth 28
Criteria for Selection of Focused Gene Panels or Comprehensive Epilepsy Gene Panels
If the epilepsy phenotyping (clinical features, EEG, neuroimaging, biochemical profile) suggests a specific broad epilepsy syndrome/group (e.g., malformations of cortical development, familial focal epilepsy, progressive myoclonic epilepsy), a focused gene panel may be indicated. On the other hand, should the clinical phenotype or laboratory markers fail to categorize the patient to a distinct epilepsy group, a comprehensive gene panel may be considered.
∙ If clinical diagnosis is clear and genetic heterogeneity is low, focused gene panels are indicated—for example, Dravet syndrome, progressive myoclonic epilepsy, cortical malformation panel, Angelman‐like syndrome and Rett‐like syndrome.
∙ If a treatable epilepsy is under consideration, a STAT epilepsy panel focused on treatable conditions should be considered. Examples of important treatable conditions include pyridoxine-dependent epilepsy, pyridoxal phosphate-dependent epilepsy, creatine deficiency syndromes, glucose transporter (GLUT1) deficiency, cerebral folate deficiency and serine biosynthesis disorders (Table 3). These are so-called “actionable gene panels.”
∙ Where the clinical diagnosis is clear and genetic heterogeneity is high, but the clinical diagnosis is not indicative of a treatable condition, either a focused gene panel or comprehensive epilepsy panel should be carried out. Examples include epileptic encephalopathies such as Ohtahara syndrome, infantile spasms, epilepsy of infancy with migrating focal seizures and Lennox Gastaut syndrome.
∙ If the clinical diagnosis is not clear and genetic heterogeneity is unknown, either a focused gene panel or comprehensive epilepsy panel should be carried out. Some of the examples are as follows:
° Seizures associated with a fever as a major trigger
° Epilepsy in females with cognitive impairments or intellectual disability
° Epilepsy syndromes associated with focal and multifocal seizures
- Autosomal dominant nocturnal frontal lobe epilepsy
- Familial temporal lobe epilepsy with auditory features
- Familial focal epilepsy with variable foci
- Sporadic Focal-onset pharmacoresistant epilepsy
° Epilepsy associated with regression/dysfunction in language/communication +/− development (Landau Kleffner syndrome, epileptic encephalopathy with continuous spike-and-wave during sleep)
° Idiopathic (genetic) generalized epilepsy refractory to treatment
∙ Epilepsies suggestive of mitochondrial diseases or in the context of mitochondrial diseases.
Table 3 Potentially treatable genetic/metabolic epilepsies

It is noteworthy that as the new epilepsy genes are being discovered and new phenotypes are being described for the known genes, the genetic results may need to be re-evaluated or genetic testing (such as focused gene panels) may need to be repeated for patients with negative or inconclusive genetic testing results.
Future Directions
In addition to the guidelines for epilepsy genetic testing described above, the Epilepsy Genetic Testing Criteria Working Group recommended future steps to maximize appropriate testing, which were supported by GTAC, and include the exploration of a role for the recently formed Ontario Epilepsy Network in promoting consultation between and among epileptologists to support optimal genetic test selection for patients, regardless of geographic location in the Province. In addition, there will be a patriation of all genetic testing for epilepsy to Ontario laboratories.
The next step, now underway in Ontario, is the creation of an epilepsy genetic testing implementation working group to develop a programmatic plan for implementing epilepsy panel testing in Ontario as a provincial service that is in alignment with the GTAC report, Criteria for Genetic Testing Related to Epilepsy. This work will lead to the creation of tailored, evidence-based epilepsy gene panels for a specific group of epileptic disorders in Ontario.
Financial Support
None.
Disclosures
The authors have nothing to disclose.
Statement of Authorship
DA, ED, DD, ANP, SG, KB, ML and OCS were part of the working group. They were involved in the study concept and design and formulating the guidelines. All authors were involved in drafting and revising the manuscript and approved the final draft being submitted for publication.





